

Le projet « Mille Génomes Gallus » : partager les données de séquences pour mieux les utiliser
Chapeau
Le séquençage du génome entier change la donne dans tous les domaines de la génétique : sélection, biodiversité, déchiffrage de la relation génotype-phénotype. Le concept d’un projet « 1000 Génomes », testé chez les bovins, consiste à mutualiser des données de séquence obtenues par différents partenaires pour faciliter l’extraction d’informations répondant à différentes questions de recherche. Les conditions de sa mise en place chez la poule sont présentées ici dans un contexte national et international.
Introduction
La poule est une espèce d’intérêt économique majeur pour l’alimentation humaine, avec 109 millions de tonnes de viande produites et 70 millions de tonnes d’œufs en 2017 (http://www.fao.org/faostat). La poule est aussi une espèce modèle utilisée de longue date pour la biologie du développement, grâce à l'accès relativement facile à l'embryon à tous les stades de développement. Cette conjonction d'espèce économique et d'espèce modèle lui a valu d'être la première espèce d'élevage à avoir son génome séquencé. L'intérêt de séquencer le génome de la poule est aussi de constituer une référence pour l'assemblage du génome d'autres oiseaux, comme par exemple la pintade, ce qui a rapidement permis d'identifier quelques gènes clés impliqués dans la domestication de cette espèce (Vignal et al., 2019).
La principale espèce ancêtre sauvage de la poule, Gallus gallus, ou encore poule de jungle, vit toujours dans son habitat naturel en Asie (Inde, Vietman, Thailande…), de même que les espèces proches que sont Gallus sonneratii, Gallus lafayetii et Gallus varius. L'étude conjointe du génome de ces espèces permet d'aborder le processus de domestication de la poule par la génomique. Le génome de la poule est plus compact que celui des mammifères, avec 1,26 milliard de bases nucléotidiques (encadré 1), soit 1,26 Gigabases (Gb) contre environ 3 Gb, son séquençage est donc moins coûteux. La première version du génome de l'espèce Gallus gallus a été obtenue à partir de l'ADN d'une lignée consanguine de poule de jungle conservée par l'Université de Californie à Davis (Hillier et al., 2004), dont le génome homozygote était plus facile à assembler de novo (encadré 1). La dernière version disponible est la version 6. Ce génome constitue un référentiel commun pour tous les travaux de génomique sur cette espèce. Il est dénommé « génome de référence ».
Encadré 1. Glossaire.
- Assemblage de novo : organisation des segments séquencés pour un individu en ensembles de segments successifs qui sont ensuite mis bout à bout pour constituer le génome entier, cette opération complexe suppose une grande quantité de séquences obtenues avec différentes méthodes.
- Bases nucléotidiques : les quatre constituants élémentaires de l’ADN : A, C, G, T.
- dbSNP : base de données internationale recensant les variants de séquence de type « Single Nucleotide Polymorphism » chez l’homme, https://www.ncbi.nlm.nih.gov/snp/
- Imputation : prédiction statistique des variants inconnus du génome d’un individu à partir d’un sous-ensemble de variants identifiés par génotypage.
- InDel : pour insertion/délétion, petite suppression (délétion) ou addition (insertion) d’un faible nombre de bases (1 à 50) par rapport au génome de référence.
- Lecture : liste des bases nucléotidiques successives lues sur un même segment du génome lors du séquençage, la longueur d’une lecture dépend de la méthode de séquençage utilisée.
- Profondeur de séquence : nombre de lectures obtenues à chaque position, ainsi une profondeur de 10X signifie que chaque position du génome est en moyenne lue 10 fois.
- Puce SNP : ensemble standardisé de marqueurs de type SNP rassemblés sur un support permettant de déterminer la base nucléotidique présente pour chaque marqueur chez chaque individu, cette opération dénommée génotypage fournit le génotype de l’individu pour cet ensemble standardisé de marqueurs, dénommé « puce de génotypage ».
- Reséquençage : séquençage du génome d’un individu n’ayant pas pour but d’assembler le génome de l’espèce mais d’identifier les variants par rapport au génome de référence.
- Signature de sélection : région du génome caractérisée par une variabilité faible, très inférieure à celle des régions voisines, en raison d’un effet de la sélection qui favorise certains variants au détriment d’autres et diminue la variabilité localement.
- Variant (génétique) : toute différence détectée entre la séquence du génome d’un individu et le génome de référence.
Une fois le génome de référence établi, le génome de nouveaux individus est séquencé sans réaliser un assemblage de novo, cette opération dénommée reséquençage (encadré 1) permet d’obtenir une masse d’informations qui peut être utilisée pour inférer l’histoire d’une espèce ou d’une race, mais aussi la diversité génétique accessible pour la sélection (Vignal et Besbes, 2005) comme pour la gestion des populations en conservation. Le génome constitue une archive de l’histoire d’une population car son analyse permet de reconstituer les variations d’effectif, de détecter de rares événements de croisements entre espèces ou entre races au sein d’une espèce, de détecter des régions du génome montrant une perte importante de variabilité en réponse à la sélection (« signature de sélection », encadré 1) ou encore d’identifier un individu ancêtre commun aux animaux porteurs d’une anomalie génétique.
Le séquençage a conduit à remplacer les marqueurs microsatellites des années 1990-2010 par les marqueurs SNP (« Single Nucleotide Polymorphism »), définis comme la variation par rapport à la séquence de référence d’une base précisément localisée dans le génome. De ce fait, les résultats de différentes études deviennent beaucoup plus facilement comparables entre eux qu’auparavant. Sur le plan technique, le séquençage a constamment gagné en efficacité et son coût a régulièrement diminué depuis les années 2000. En 2019, le coût du séquençage d’un génome entier de poule, avec une profondeur de 10X (encadré 1) est d’environ 300 euros, parfois moins, selon les prestataires de séquençage. Le résultat de ce séquençage comprend toutes les variations présentes chez l’individu par comparaison au génome de référence, et permet ensuite d’identifier celles qui lui sont propres. À titre d’exemple, le génotypage d’une poule par le seul outil public à haute densité de 580 000 marqueurs SNP coûtait environ 140 euros en 2019 pour produire une information certes très utile mais ne permettant pas de détecter les variants nouveaux propres à l’individu étudié.
Toutefois, l’extraction des informations pertinentes à partir des données de reséquençage suppose un travail d’analyse beaucoup plus important que celui nécessaire pour exploiter les résultats d’une puce de génotypage. Les principales étapes sont résumées dans la figure 1. Identifier un variant génétique (encadré 1) par sa position en nombre de bases sur un chromosome permet aussi de rechercher dans les bases de données si d’autres variants ont déjà été décrits à cette position ou à son voisinage.
L’analyse de données devient le facteur limitant, mutualiser l’analyse permet donc de mieux exploiter les données de reséquençage et de génotypage, et d’augmenter la puissance des comparaisons entre individus ou entre groupes : c’est la principale motivation d’un projet « Mille Génomes ». L’objectif de cet article est de décrire le contexte conduisant à lancer un projet « Mille Génomes », d’expliquer les objectifs d’un tel projet dans le cas de l’espèce Gallus gallus, de présenter les premiers résultats d’un projet pilote conduit sur les données de séquence obtenues en France, et d’analyser les possibilités d’ouverture internationale de ce projet.
1. Contexte de l’open data et de l’open science
Les agences de financement de la recherche et notamment l’Union Européenne ont souhaité que les données produites grâce à des financements publics soient accessibles à tous les acteurs de la recherche, publics ou privés, et plus généralement à tous les citoyens, qui financent indirectement ces recherches. Cette stratégie dite open data est considérée comme nécessaire au développement de l’innovation à partir de la recherche. Elle s’inscrit dans une volonté plus large de science ouverte à la société. Elle est particulièrement relayée par la « Research Data Alliance » (https://www.rd-alliance.org/) dont le nœud français est coordonné par le CNRS dans le cadre du projet européen RDA Europe 4.0, qui a démarré le 1er mars 2018. Les institutions de recherche, dont INRAE, ont adhéré à cette stratégie et accompagnent leurs chercheurs dans cette démarche non intuitive, chaque chercheur ayant naturellement le réflexe de conserver ses données pour traiter sa question. Il ne s’agit pas de diffuser n’importe comment des données mais de les produire et de les conserver de manière standardisée, en les documentant avec des « métadonnées » utilisant un vocabulaire standardisé afin de faciliter leur réutilisation. L’idée est bien qu’une seule personne ou qu’un seul groupe de chercheurs ne peut exploiter totalement un jeu de données et que le partage de ces données améliore leur utilisation et la connaissance qui en est extraite. L’enjeu est aussi de ne pas être le dernier à exploiter ses données et la stratégie open data pousse à une certaine accélération de la recherche. Ainsi, chaque projet de recherche doit comporter un plan de gestion des données, décrivant le type de données produites, leurs conditions d’accès et d’utilisation futures (stockage, période d’embargo, reconnaissance du producteur initial des données).
La génomique se prête particulièrement bien à cette stratégie de partage des données, grâce à la standardisation de la description des données de séquence et à l’existence d’entrepôts internationaux qui rassemblent les données du génome de référence et des génomes individuels, tels que NCBI développé aux États-Unis (https://www.ncbi.nlm.nih.gov/), Ensembl développé en Europe (http://www.ensembl.org) ou encore UCSC développé par une université américaine (https://genome.ucsc.edu/).
L’accès aux échantillons devient un enjeu associé à l’open data : la connaissance qui peut être extraite des données de séquence dépend aussi des informations attachées aux échantillons. Certaines institutions, comme la China National Gene Bank, fondent leur partenariat sur une offre de séquençage à bas coût, qui leur permet de collecter des échantillons sans envoyer les leurs. Or, l’accès aux échantillons est encadré par le protocole de Nagoya pour l’accès aux ressources génétiques et le partage des avantages retirés de leur utilisation
2. Le concept « Mille Génomes »
Le concept « Mille Génomes » a été inventé pour l’espèce humaine (www.internationalgenome.org) en 2010, avec une publication de référence en 2012 décrivant la variabilité génétique de 1092 génomes humains (The Mille Genomes Project Consortium, 2012). L’espèce bovine a été la première espèce domestique à bénéficier de cette approche et constitue l’exemple de référence pour mettre en place un projet « Mille Génomes » pour la poule. L’objectif général de ce type de projets est d’augmenter la puissance des analyses génomiques en augmentant le nombre de génomes séquencés accessibles pour une même analyse.
2.1 Un précédent dans les espèces d’élevage : le projet « Mille Génomes bovins »
L'idée est née en Australie en 2011 et un premier consortium a été créé en 2012 avec 7 partenaires dont INRAE. L'objectif de ce premier projet était d'avoir accès à un grand nombre de séquences individuelles sans augmenter les coûts par partenaire, au sein d'un consortium dans le cadre d'un contrat signé par chaque membre. Pour un membre, l'intérêt est double : il accède à une quantité d'information beaucoup plus importante ; il bénéficie d'un traitement des données normalisé. Les lectures une fois alignées sur le génome de référence (figure 1- étape 1) sont rassemblées dans un même site où sont réalisées toutes les étapes de traitement avec une même méthode, afin de redistribuer à tous les membres du consortium les résultats de l'étape 3 (figure 1). La première publication de référence issue de ce projet date de 2014 et montre que l'analyse de la séquence de 234 taureaux facilite la cartographie génétique de caractères monogéniques mais aussi de caractères complexes (Daetwyler et al., 2014).
Figure 1. Analyse des données de séquençage du génome entier utilisées pour un projet « Mille Génomes ».
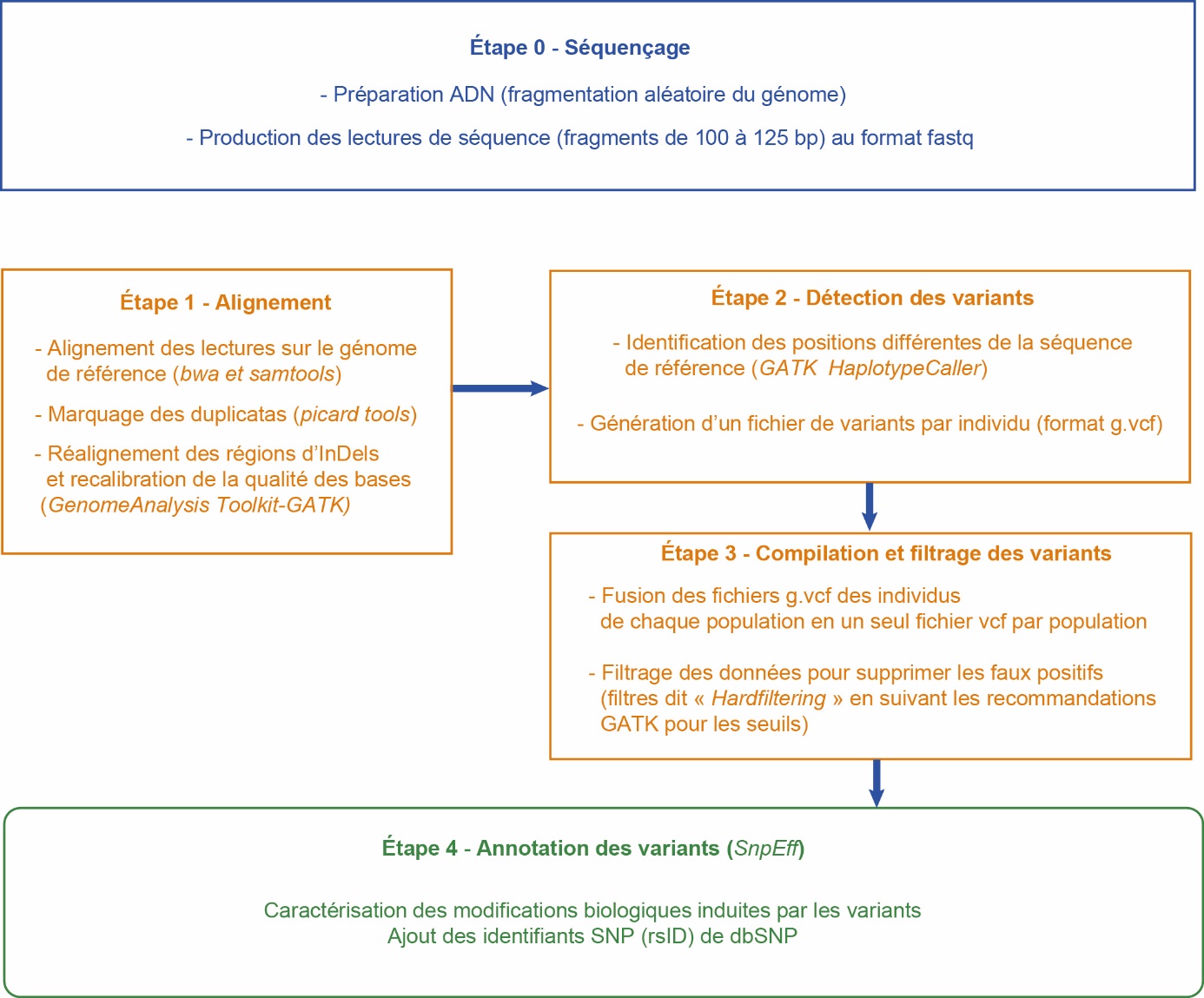
Après le séquençage, l’étape 1 identifie les fragments obtenus en les alignant sur le génome de référence. L’étape 2 compare les fragments localisés à la séquence de référence pour en identifier les variations. L’étape 3 applique différents filtres pour éliminer les faux positifs. L’étape 4 (facultative) est l’annotation fonctionnelle des variants. Les noms des outils bioinformatiques dédiés sont indiqués en italique. L’acronyme dbSNP est défini dans l’encadré 1.
Pour garantir l’homogénéité du traitement, seules les lectures de séquences obtenues par la technologie Illumina sont partagées et caractérisées par des descripteurs minimaux obligatoires : race, identifiant de l’animal (potentiellement rendu anonyme mais unique), sexe, profondeur de séquence, plusieurs critères techniques de réalisation du séquençage. Les membres du consortium sont obligatoirement apporteurs de données de séquences : à la création du consortium, le minimum d’apport était d’au moins 10 animaux avec une profondeur individuelle au moins égale à 4X pour un total supérieur à 100X (par exemple, au moins 10 animaux à 10X ou 25 animaux à 4X). Les données ont été apportées par fournées successives, dénommées « run » : de 125 animaux au « run » 1, à 3100 au « run » 7. L’apport minimal demandé augmente à chaque « run » afin de renforcer l’engagement des partenaires. Le consortium comptait plus de 30 membres en 2017. Environ la moitié des données de séquences brutes sont déposées dans les sites publics au fur et à mesure des publications réalisées. Le choix du dépôt en base publique relève de l’apporteur de données et n’est pas une obligation du consortium. On voit donc ici un exemple où l’open data est contrôlé : les données sont d’abord partagées au sein d’un consortium avant d’être progressivement rendues publiques.
Dans ce consortium, chaque membre finance ses coûts, y compris de séquençage. Le coordinateur australien (Agriculture Victoria Research, Melbourne, Australie) finance le stockage des données et les analyses communes (étapes 1 à 3, figure 1), alors que le travail d’annotation (étape 4, figure 1) est partagé entre différents membres du consortium. En 2019, le « run » 7 a permis d’identifier 90 millions de SNP et 13,5 millions d’InDels à partir des données de séquence fournies par 40 partenaires sur 3100 individus de l’espèce Bos taurus issus de 106 races. Une particularité de ce consortium est aussi d’améliorer la qualité des données apportées en utilisant l’information de déséquilibre de liaison à l’échelle de l’échantillon total pour fournir la probabilité a posteriori du génotype pour chaque variant connu pour chaque individu. Ce traitement conduit à inférer les génotypes absents ou à corriger les génotypes peu fiables et à faire disparaître les pseudo-allèles issus d’erreurs de séquençage. Cette étape importante pour la qualité des résultats n’est possible qu’en traitant l’ensemble des données, elle constitue donc une plus-value du consortium.
Il est capital de noter que les phénotypes et les génotypes utilisés en sélection ne sont pas partagés. Chaque partenaire peut réaliser pour son compte des analyses avec les données mutualisées. Ainsi, les associations entre variants de séquence (SNP + InDels) et phénotypes sont réalisées par les partenaires sur leurs propres données de phénotypes et ces données sont partagées lorsque l'ensemble des membres du consortium le jugent utile. Cela a permis au consortium d'identifier l'ensemble des gènes sous-tendant les locus de caractères quantitatifs (QTL) déterminant la taille des bovins et d'en faire une publication commune (Bouwman et al., 2018).
Une autre valorisation importante pour la sélection est de pouvoir reconstituer la séquence d’un animal qui n’a été que génotypé. Ainsi, la connaissance du génotype à un certain nombre de positions dans le génome (en général au moins 50 000) permet de déduire la séquence la plus probable entre chaque position génotypée grâce au référentiel de SNP établi par le projet « Mille Génomes » pour des individus des races représentées dans le jeu de données. De ce fait, des animaux génotypés sont presque aussi bien connus que des animaux séquencés dans une race donnée. Cette étape s’appelle « imputation » (encadré 1) et peut être réalisée par un partenaire sans avoir à partager les résultats obtenus. Elle améliore grandement la précision de la sélection conduite par ce partenaire et permet aussi de construire de grandes populations de cartographie pour identifier directement les variants candidats sous-jacents aux QTL. Elle est d’autant plus utile et précise que le nombre d’animaux génotypés et apparentés aux animaux séquencés est grand. Ainsi, en France et à ce jour, près de 100 000 animaux avec phénotypes et génotypes SNP de puces ont été imputés à l’échelle de la séquence. Cet argument d’imputation a été décisif pour constituer le consortium bovin à l’époque où la sélection génomique se mettait en place dans tous les pays. La majorité des animaux séquencés correspondent à des taureaux ancêtres des populations de production actuelles, ayant une forte contribution génétique à leur population. Ils sont très informatifs en termes de variants présents dans les populations mais n’ont plus aucun intérêt stratégique en termes de phénotypes ou de sélection.
2.2 Objectifs du projet « Mille Génomes Gallus »
Partant du même objectif général présenté pour l’espèce humaine et pour l’espèce bovine, le projet proposé pour la poule développe quatre types d’applications.
a. Analyse de la structure du génome
Le génome de la poule est non seulement plus compact que celui des mammifères, mais son organisation se caractérise par l’existence de quelques très grands chromosomes, dénommés macrochromosomes, et de nombreux petits chromosomes, dénommés microchromosomes, qui constituent une particularité des oiseaux (à l’exclusion des rapaces).
Rassembler le plus grand nombre de séquences permettra non seulement une identification plus complète des variants de type SNP et InDel, mais aussi d’étudier leur répartition entre macrochromosomes et microchromosomes, qui sont particulièrement difficiles à analyser.
Une étape supplémentaire par rapport au projet bovin serait aussi de répertorier l’ensemble des variants structuraux, par exemple les insertions rétrovirales qui ont été étudiées chez la poule dès les années 80, et dont plusieurs ont été associées à des modifications morphologiques majeures (coquille bleue, plumage blanc, emplumement lent…). Ce phénomène est également décrit chez d’autres espèces, dont les bovins, mais de nombreuses races de poule ont fait l’objet d’études systématiques des insertions rétrovirales dans les années 90, sans que l’ensemble de ces insertions ait été véritablement catalogué, ce que permettrait le projet « Mille Génomes ».
b. Caractérisation de la diversité de l’espèce
La poule est une espèce à intervalle de génération court, qui a vu un grand nombre de races se différencier en quelques siècles, qui a accumulé le plus grand nombre de générations de sélection pour des populations d’élevage commerciales, et qui a aussi permis le développement d’un grand nombre de lignées expérimentales présentant des phénotypes extrêmes. On peut ainsi identifier 3 groupes principaux de populations :
i) les lignées commerciales de poules pondeuses : on distingue les poules pondeuses à œufs blancs, toutes dérivées de la race Leghorn blanche, et les poules pondeuses à œufs bruns, dérivées de quelques races dont la « Rhode Island Red » (RIR) et la « New Hampshire » ;
ii) les lignées commerciales de poulets de chair : on distingue les lignées à croissance très rapide, dérivées de quelques races comme la « Cornish » et la « White Plymouth Rock », et les lignées à croissance modérée, essentiellement représentées par les poulets dits ‘label’ développés en France depuis les années 1960 à partir de populations locales, notamment du Sud-Ouest de la France, ayant une diffusion internationale, bien inférieure à celle des lignées à croissance très rapide, mais significative ;
iii) les races locales : la plupart sont des races conservées en petits élevages par des éleveurs amateurs sur la base de critères morphologiques, mais certaines races françaises sont engagées dans des programmes de valorisation pour la production de viande de qualité, telles que le poulet de Bresse.
Il n’existe donc pas une seule race aussi présente à l’échelle internationale que l’est la race bovine Holstein, et l’analyse de la diversité génétique est un objectif sans doute plus important que pour le projet « Mille Génomes bovin ». De plus, les différentes puces de génotypage utilisées chez la poule ont généralement peu de marqueurs en commun, car elles ont été mises au point à partir de certaines lignées commerciales et rarement de races locales, ce qui empêche la mutualisation de ces données, alors que le séquençage permettrait de les connecter.
Les applications envisagées concernent l’identification d’allèles spécifiques de races ou de lignées, l’étude de scénarios de domestication et la détection de flux de gènes entre populations sauvages et domestiques ou entre populations domestiques, par exemple entre races locales et lignées commerciales, voire entre lignées commerciales. La condition est évidemment que des données de séquence soient apportées pour ces différents types de populations.
c. Identification de mutations causales
La connaissance de la séquence permet d’accéder à l’ensemble des variants chez un individu, alors que les données de génotypage ne permettent d’identifier que le génotype pour des marqueurs déjà connus. L’intérêt de rassembler les données de séquence est donc de pouvoir identifier les variants nouveaux susceptibles de déterminer un phénotype particulier. La connaissance de ces mutations causales permet, d’une part, de mieux comprendre le fonctionnement des gènes qui les portent, et, d’autre part, d’ajouter ces variants sur les puces de génotypage ou de développer des tests de diagnostic moléculaire ciblés si le génotypage sur puce est trop coûteux.
Plusieurs cas de figure peuvent se présenter :
i) répertorier toutes les variations possibles pour un gène de fonction connue et rechercher les variants spécifiques à une population, susceptibles d’être associés à un phénotype propre à celle-ci. Les partenaires disposant de données de phénotypage sur les animaux séquencés pourraient alors progresser dans l’analyse du lien phénotype-génotype ;
ii) identifier la base génétique d’un phénotype à déterminisme mendélien en comparant des sous-ensembles de populations définis sur la base de ce phénotype, afin de détecter des fréquences alléliques contrastées entre ces sous-ensembles ; la constitution des sous-ensembles suppose de connaitre le phénotype des individus ou de la population si elle est est homogène pour ce caractère, et donc d’avoir l’accord des fournisseurs de données ;
iii) rechercher des signatures de sélection entre deux lignées divergentes : cette analyse sera généralement prise en charge par le partenaire ayant produit les lignées mais ce partenaire aura la possibilité de savoir si les variants candidats trouvés dans ses lignées sont uniques ou plus ou moins fréquents dans d’autres populations.
La recherche de déficit en homozygotes comme cela a pu être fait chez les bovins est limitée chez la poule par le nombre trop faible de génotypes disponibles et partageables pour une race donnée. Ce type d’analyse sera plutôt réalisé par chaque sélectionneur sur données privées.
d. Imputation des variants inconnus du génome à partir des génotypes
Comme dans le cas du projet « Mille Génomes bovin », mutualiser les données des animaux séquencés permettra de mieux tirer parti des données privées de génotypage des candidats à la sélection. Le partage des séquences améliorera l’analyse des dispositifs privés de chaque membre du consortium de séquençage, et notamment ceux des partenaires privés, sans qu’il leur soit besoin de partager leurs données de génotypage ou de phénotypage. Cette application s’est révélée particulièrement utile pour la détection d’anomalies létales à l’état homozygote chez les bovins, en enrichissant les nombreuses données de génotypage par la prédiction de la séquence. Toutefois, actuellement, on constate que chaque sélectionneur avicole préfère développer sa propre règle d’imputation, pour des raisons de compétition exacerbée, ce qui limite l’intérêt pour eux à participer à un projet de partage de données du type « Mille Génomes ».
3. Le projet pilote français « Mille Génomes Gallus »
3.1. Origine des données
Ce pilote s’appuie sur la mutualisation des données de séquence de 207 animaux issues de neuf projets de recherche financés par l’Agence Nationale de la Recherche ou l’Europe, et conduits par des équipes françaises sur des races locales non sélectionnées, des lignées expérimentales ou commerciales, de type ponte ou de type chair (tableau 1). Chaque projet devient en quelque sorte un témoin pour les autres. Les animaux séquencés proviennent des 4 espèces du genre Gallus, si bien qu’on peut parler d’un projet « Mille Génomes Gallus ».
Tableau 1. Description des jeux de données individuelles rassemblés dans le projet pilote « Mille Génomes Gallus », en fonction du projet de recherche, du type de populations étudiées et de la profondeur de séquence moyenne.
Projet |
Correspondant |
Type de population |
Nombre |
Profondeur |
|---|---|---|---|---|
ACRIGEN |
E. Le Bihan-Duval |
Poulet de chair à croissance rapide, |
16 |
18 |
CHICKSEQ |
S. Lagarrigue |
Poulet de chair à croissance rapide, |
24 |
15 |
Domestichick |
M. Tixier-Boichard |
Races locales (tous les continents : |
36 |
33 |
EpiBird |
F. Pitel |
Race Leghorn blanche pondeuse à œufs blancs Race Rhode Island Red (RIR), |
2 |
15 |
Feed-a-Gene |
S. Lagarrigue |
Race RIR, lignée expérimentale |
19 |
46 |
FR-AgEncode |
E. Giuffra (GABI) |
Race Leghorn blanche, lignée expérimentale |
4 |
38 |
SABRE |
S. Mignon-Grasteau |
Poulet de chair de type label issu d’un |
6 |
29 |
UtOpIGe |
P. Le Roy (PEGASE) |
Lignée commerciale de pondeuses |
90 |
14 |
Données |
Races locales d’origine asiatique, |
88 |
20 |
|
Total |
295 |
Chaque jeu de données (défini pour chaque projet) est décrit par un ensemble de 18 métadonnées (tableau 2). L’ensemble de ces données est regroupé sur le serveur de la plateforme SIGENAE du département de Génétique Animale de INRAE à Toulouse.
Tableau 2. Métadonnées utilisées pour décrire les jeux de données
Catégorie |
Métadonnée |
|---|---|
Jeu de données |
Nom du projet |
Population |
Espèce |
Statut : domestique ou sauvage |
|
Type : non sélectionnée, locale, commerciale, |
|
Nom : standardisation à mettre en place |
|
Individu |
Identifiant : format à standardiser |
Sexe |
|
Pedigree : oui/non |
|
Identifiant_père |
|
Identifiant_mère |
|
Echantillon ADN |
Mélange : oui/non |
Nombre d'individus/ mélange |
|
Séquençage |
Technologie |
Date d'obtention des données |
|
Longueur des fragments en bp |
|
Méthode paired-end : oui / non |
|
Profondeur : nombre de génomes-équivalents |
|
Code md5 : code obtenu à l’issue |
L’exploration des bases de données publiques a permis d’identifier 88 données supplémentaires de séquences individuelles disponibles pour la poule, qui représentent principalement des races locales asiatiques (67 individus), quelques lignées commerciales (6) ou expérimentales (9) et 6 individus sauvages. On peut donc déjà disposer de 295 séquences individuelles ; toutefois la profondeur des séquences varie de 5 à 90X. En considérant une profondeur minimale de 10X pour garantir une information assez fiable, on doit retirer 20 séquences individuelles, dont 4 issues du projet pilote français. Il reste alors un ensemble de données individuelles de bonne qualité pour 275 animaux, soit du même ordre de grandeur que le « run » 2 du projet bovin.
Par ailleurs, un projet, dénommé QTLDJ, financé par le département de Génétique Animale d’INRAE, a permis de séquencer quatre mélanges d’individus de lignées expérimentales de race Leghorn blanche, se distinguant par le taux d’ovulations multiples. Les séquences obtenues sur mélange d’ADN ne sont pas la cible prioritaire du projet, mais peuvent être utilisées pour des questions spécifiques.
3.2 Méthodes d’analyse
L’étape 1 (figure 1) a été réalisée pour les 207 séquences individuelles des projets français en utilisant la version 5 du génome de référence de Gallus gallus, mais la version 6 étant disponible depuis 2019, il est probable qu’un nouvel alignement sera nécessaire dans le futur. Les étapes 2 à 4 ont été réalisées comme décrit dans la figure 1.
a. Analyse globale de la diversité
Nous avons commencé par des analyses de diversité génétique pour illustrer l’intérêt de la mutualisation des données. Pour cela, nous avons conservé uniquement les SNP pour lesquels un identifiant était disponible dans la base dbSNP (environ 5 millions de SNP concernés). La matrice des distances génomiques entre individus deux à deux a été calculée avec le logiciel plink (version 1.9) sur la base de l’identité par état (Identity by State, IBS) des allèles à chaque locus. Ces distances ont ensuite été utilisées pour obtenir une représentation graphique multidimensionnelle (MDS) de la diversité des échantillons.
b. Structuration de la diversité génétique
Les analyses de structure de population ont été réalisées avec le logiciel ADMIXTURE (Alexander et al., 2009). Le nombre de groupes K (c'est-à-dire le nombre de populations ancestrales) a été estimé en utilisant la procédure de validation croisée disponible avec ADMIXTURE (option --cv) en faisant varier K de 1 à 30 et en choisissant la valeur de K permettant de minimiser l'erreur de validation croisée.
c. Recherche de mutations causales
Ce type d’analyses relève de l’étape 4 (figure 1). Il consiste à prédire l’impact fonctionnel d’un variant en fonction de sa localisation dans la séquence. Un SNP ou un InDel peuvent modifier l’expression d’un gène et/ou modifier la séquence de la protéine produite. Un InDel peut supprimer ou créer un codon stop ou décaler le cadre de lecture des codons, ce qui conduit à une protéine tronquée ou allongée ou de séquence d’acides aminés anormale. Enfin, un SNP dans une séquence codante peut modifier un acide aminé, avec des conséquences fonctionnelles plus ou moins sévères selon le rôle de cet acide aminé dans la protéine. Différents outils existent pour prédire l’effet plus ou moins délétère des variants recensés : l’outil « Variant Effect Predictor » (VEP) développé par l’« European Bioinformatics Institute » (EBI) est le plus accessible. Il est souvent utile de tenir compte de la conservation de la séquence entre espèces : un haut niveau de conservation indique que la séquence est sous sélection et doit affecter une fonction importante, voire vitale. Tout variant dans une séquence très conservée a une forte probabilité d’avoir un effet délétère. Cette analyse de variants a été appliquée à deux situations : i) la recherche de variants dans des gènes connus pour être impliqués dans une fonction d’intérêt (coloration du plumage ou de la coquille), ii) la recherche de variants délétères dans des régions où sont localisés des QTL afin d’identifier des mutations causales.
Le gène MC1R, gène de coloration des plumes, a été choisi pour valider la démarche, en tant que gène candidat pour lequel plusieurs variants ont déjà été publiés (Ling et al., 2003). Pour définir les positions de la séquence codante du gène sur la séquence génomique, nous avons comparé avec l'outil blastn cette séquence codante (NM_001031462) et la séquence génomique du chromosome 11 (NC_006098.4) connu pour porter MC1R. Puis, pour chaque population, la région génomique contenant la séquence codante (un seul exon) du gène MC1R sur le chromosome 11 (assemblage GalGal5 : NC_006098.4 - 11:19084582-19085526) a été extraite des fichiers issus de l'étape 4. Les fréquences alléliques ont été calculées avec le logiciel vcftools. La position des variants ainsi identifiés a été comparée aux positions des mutations déjà connues.
Cette stratégie a ensuite été appliquée à des gènes connus comme impliqués dans une voie métabolique cible mais pour lesquels les mutations restent à découvrir. Une liste de sept gènes impliqués dans le métabolisme de la protoporphyrine IX, principal pigment déterminant la couleur brune de l'œuf, a été établie à partir de la littérature (Samiullah et al., 2015 ; Li et al., 2013) : ABCG2, ABCB6, ALAS1, CPOX, FECH, FLVCR1, SLC25A38. Il s'agit de rechercher les SNP de ces gènes montrant une fréquence opposée (0 ou 1) entre des lignées de race Leghorn blanche, qui pondent des œufs blancs (projets FR-Agencode, et QTLDJ) et des lignées de race Rhode Island Red, qui pondent des œufs bruns (projets UtOpIGe et Feed-a-Gene).
Finalement, des régions QTL contrôlant la qualité de l'œuf (Romé et al., 2015) ont été choisies pour rechercher les variants délétères dans le sous-ensemble de 12 millions de SNP identifiés chez 175 individus de types génétiques « commerciaux » (chair ou ponte).
3.3 Résultats
a. Analyse globale de la diversité
Le nombre total de SNP détectés atteint 42 millions. Les jeux de données montrant les valeurs les plus faibles (moins de 10 millions de SNP) correspondent aux projets qui comprennent peu d’animaux (Fr-AgEncode, EpiBird) ou des animaux de lignées fermées ou sélectionnées depuis un grand nombre de générations (CHICKSEQ, UtOpIGe). Les poulets de chair et les poulets labels montrent plus de variation que les poules pondeuses et que les lignées expérimentales, alors que les individus sauvages Gallus gallus montrent la plus grande variation (14 millions). Les 3 autres espèces sauvages montrent aussi un haut niveau de variation par rapport au génome de référence Gallus gallus, avec 12 à 14 millions de SNP.
L’analyse des distances génétiques IBS et leur représentation par graphique MDS (Figure 2) permettent de visualiser la gamme de variation présente dans le jeu de données. Le 1er axe montre la dispersion des populations selon leur niveau de variabilité : à gauche les poules sauvages et traditionnelles, plus variables (voir paragraphe 3.3.a), puis les poulets de chair et enfin les pondeuses à droite, moins variables. L’axe 2 sépare les pondeuses à œufs bruns commerciales (UtOpIGe) en haut à droite des pondeuses à œufs bruns expérimentales en bas à droite, avec un gradient allant des lignées expérimentales de pondeuses à œufs blancs et de poulets de chair jusqu’aux races locales. Les données du projet UtOpIGe montrent des distances génétiques faibles entre individus.
Figure 2. Projection des individus en fonction des distances génétiques d’identité par état (IBS).
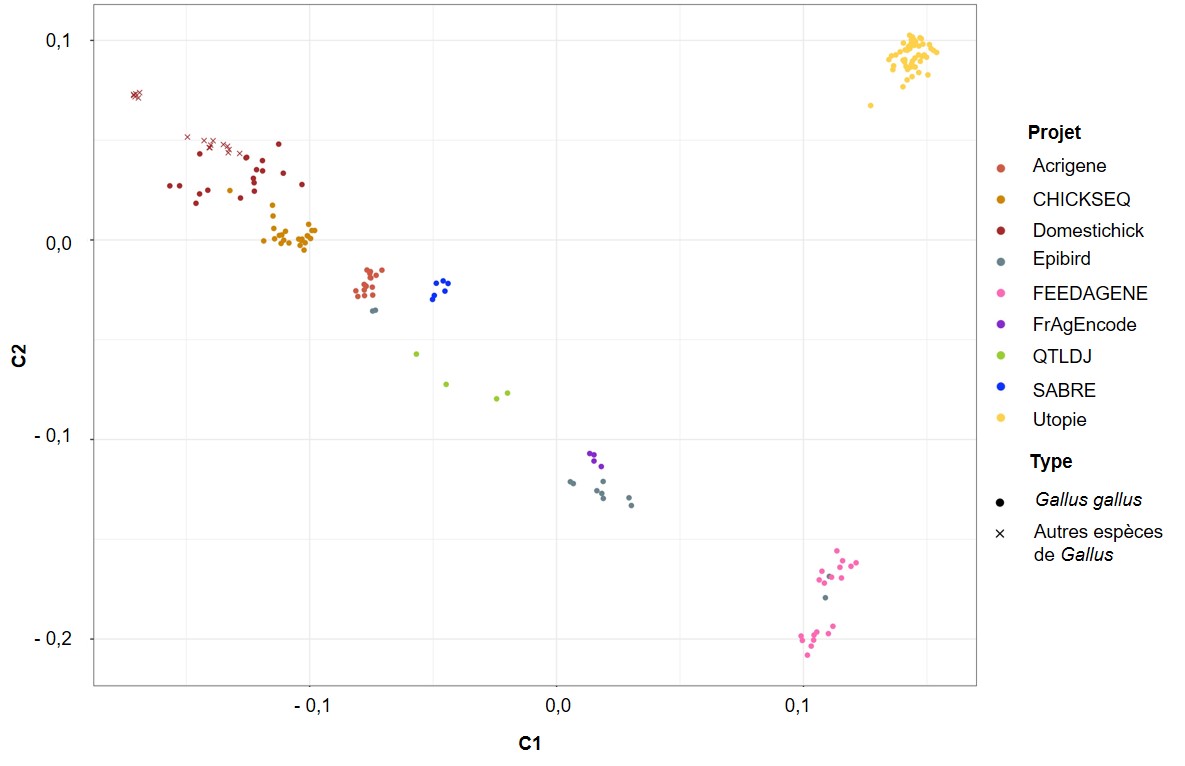
Le symbole ‘x’ désigne les individus sauvages du projet Domestichick.
b. Structuration de la diversité génétique
L’analyse de structure de population fait apparaitre sept regroupements ou clusters (figure 3). Le cluster 4 est défini par le projet ACRIGEN (poulet de chair moderne) et le cluster 5 par le projet UtOpiGe (pondeuses œufs bruns) alors que le cluster 2 est caractéristique du projet ChickSeq (lignée ancienne de poulet de chair), mais on détecte un apparentement faible avec d’autres clusters à l’intérieur de ce groupe. Les animaux sauvages Gallus gallus définissent le cluster 7 et ceux des 3 autres espèces le cluster 6. Les races locales ont un fort apparentement au cluster 7 et montrent un apparentement faible à modéré avec tous les autres clusters, y compris le cluster 6, ce qui traduit l’absence de sélection forte chez ces populations qui sont ainsi plus proches de l’ancêtre sauvage non sélectionné. Le cluster 3 correspond probablement à l’origine White Leghorn présente dans les projets Epibird et Fr-AgEncode. Enfin, le projet SABRE, constitué d’animaux croisés F1 entre des lignées de poulet label, montre logiquement une composition diversifiée du génome avec un apparentement à 6 clusters, assez proche de la situation observée pour les races locales. Il est possible d’identifier quelques régions du génome qui sont encore partagées entre des races locales et des races très sélectionnées alors que les races très sélectionnées ont définitivement perdu une fraction de la diversité génomique de l’espèce Gallus et sont les plus éloignées de l’ancêtre sauvage.
Figure 3. Résultats de l’analyse de structure montrant l’existence de 7 groupes génétiques différents (clusters).
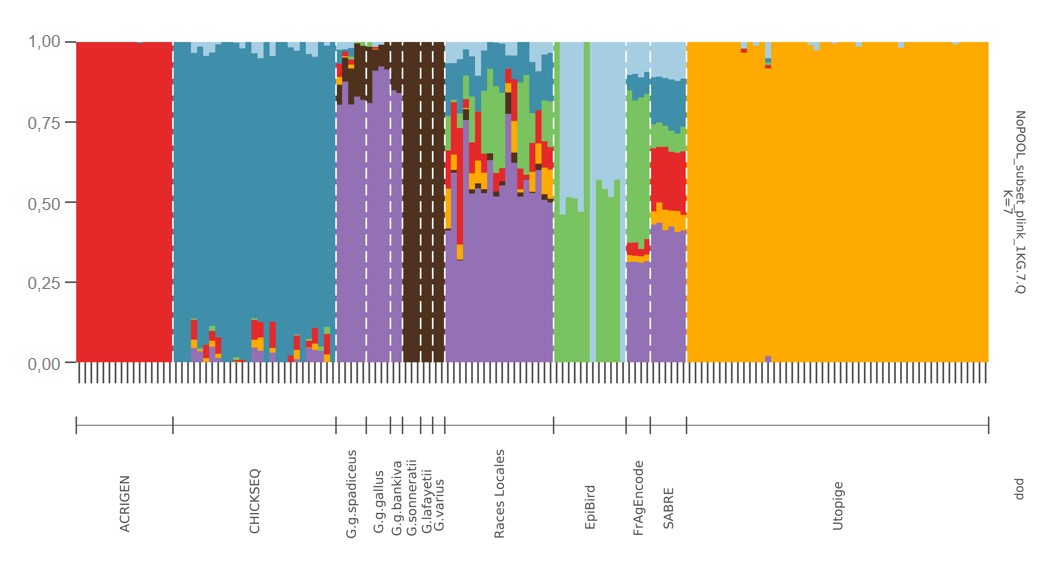
Un animal est représenté par une colonne, les segments de couleur différente dans une même colonne représentent la proportion du génome de l’individu appartenant aux différents groupes génétiques, chacun étant associé à une couleur.
c. Recherche de mutations causales
La séquence codante de MC1R comporte un exon pour lequel 10 SNPs ont été identifiés entre les populations étudiées (Tableau 3). On observe nettement la fixation de l'allèle variant en position 427 chez les pondeuses à œufs bruns qui sont les seules à avoir un plumage coloré rouge alors que les variants en position 636 et 637 sont fixés chez toutes les pondeuses et en ségrégation chez les poulets de chair. Le SNP en position 427 correspond au remplacement d'une thréonine par une alanine en position 143 du récepteur et a déjà été associé à la couleur rouge (Ling et al., 2003).
Tableau 3. Fréquence des variants de type SNP trouvés dans la séquence codante du gène MC1R. Une valeur 0 indique la présence homozygote de l’allèle de la séquence de référence.
Position |
UtOpIGe |
Feed-a-Gene |
FR-AgEncode |
ACRIGEN |
CHICKSEQ |
SABRE |
|---|---|---|---|---|---|---|
Pondeuses œufs bruns |
Leghorn blanche |
Poulets de chair |
||||
69 |
0 |
0 |
0 |
0,75 |
0,35 |
0,5 |
212 |
0 |
0 |
0 |
0,75 |
0,34 |
0,5 |
274 |
0 |
0 |
0,88 |
0,75 |
0,53 |
0,5 |
376 |
0 |
0 |
0,88 |
0 |
0,07 |
0 |
398 |
0 |
0 |
0,13 |
0,25 |
0 |
0,33 |
427 |
1 |
1 |
0 |
0 |
0,41 |
0,17 |
636 |
1 |
1 |
1 |
0,25 |
0,475 |
0,5 |
637 |
1 |
1 |
1 |
0,25 |
0,475 |
0,5 |
644 |
0 |
0 |
0 |
0,03 |
0,07 |
0,5 |
834 |
0 |
0 |
0,13 |
0 |
0 |
0 |
Le variant en position 376 est le plus fréquent en Leghorn blanche et présent chez quelques poulets de chair du projet CHICKSEQ. La variant 644 est le plus fréquent chez les poulets labels du projet SABRE. Les poulets de chair ne sont jamais fixés pour aucun variant de MC1R, ce qui est cohérent avec le fait qu’ils ont été sélectionnés pour un plumage blanc, dont le déterminisme génétique ne fait pas intervenir MC1R, alors que les pondeuses à œufs bruns ont été sélectionnées pour une couleur de plumage rouge, sur lequel MC1R joue un rôle important. La situation des poules Leghorn blanche est intermédiaire car les variants 636 et 637 sont présents chez les 4 animaux étudiés, un plus grand nombre d’individus devraient être génotypés pour confirmer la fixation de ces variants dans cette population.
En ce qui concerne les sept gènes impliqués dans le métabolisme de la protoporphyrine IX, deux d’entre eux (ABCG2 et SLC25A38) n’ont montré aucun polymorphisme contrasté entre pondeuses à œufs blancs et pondeuses à œufs bruns, et 57 variants ont été identifiés sur les cinq autres gènes. Deux gènes portent la plus grande partie des variants : FECH et ABCB6. FECH code pour l’enzyme qui catalyse l’étape finale de conversion de la protoporphyrine IX en hème, alors que ABCB6 intervient plus en amont et code pour un transporteur du coproporphyrinogène III du cytosol vers la mitochondrie, qui entre ensuite dans la chaine de synthèse de la protoporphyrine IX. FECH (sur le chromosome Z) présente 38 SNP sur un segment de 14kb, pour lesquels des allèles alternatifs sont fixés en fonction du type de pondeuses, mais la plupart sont des variants localisés en région non-codante dont l’effet fonctionnel n’est en général pas connu et difficile à prédire. ABCB6 (sur le chromosome 7) présente 7 SNP de fréquences contrastées, sur un segment de 7kb, dont un des variants est non-synonyme (il modifie un acide aminé de la protéine) avec un effet prédit qualifié de ‘MODERATE’, ce qui veut dire que la protéine n’a pas totalement perdu sa fonction. Ces deux gènes doivent maintenant être étudiés dans d’autres populations et l’effet des variants observés sur la fonctionnalité de la protéine codée doit être précisé.
En ce qui concerne la recherche de mutations sous-tendant des QTLs, un total de 3083 polymorphismes « délétères » impactant 917 gènes a été identifié pour 44 régions QTL impliquées dans la qualité de l'œuf chez des pondeuses à œufs bruns et couvrant un total de 28,6 Mb (Romé et al., 2015). Dans cet ensemble, 248 SNPs sont prédits comme ayant un impact fort sur le produit de 173 gènes et 34 polymorphismes affectent les codons start ou stop de 32 gènes. Une liste de 255 polymorphismes impactant 46 gènes a été retenue pour génotypage et analyse. Ces polymorphismes correspondent à 240 SNP et 15 InDel, et sont répartis sur 17 chromosomes. La majorité des polymorphismes suivis correspond à des substitutions d'acide aminé, 4 correspondent à des décalages de cadre de lecture, 7 modifient le cadre d'épissage et un variant provoque la création d'un codon stop. À partir d'un génotypage ciblé de ces 255 polymorphismes sur 8 lignées de poules pondeuses (4 lignées Rhode-Island Red, 4 lignées Leghorn blanche, 60 poules par lignée), nous pourrons estimer la fréquence de chaque polymorphisme dans ces différentes lignées et estimer son effet sur les caractères de qualité de la coquille.
On voit donc que la comparaison des données de séquence permet de cibler les polymorphismes à analyser plus en détails afin d’identifier, puis de confirmer la présence d’une mutation causale. On peut ajouter que les polymorphismes trouvés en opposition de fréquence entre les pondeuses Leghorn blanche à œufs blancs et les pondeuses Rhode Island Red à œuf bruns ne sont pas les mêmes que ceux détectés dans les régions QTLs des pondeuses à œufs bruns, ce qui n’est pas étonnant car la variabilité de la coloration de la coquille à l’intérieur d’une lignée (donnant lieu aux QTLs détectés) n’est pas du même ordre que la différence extrême notée entre ‘œuf blanc Leghorn’ et ‘œuf brun RIR’.
4. Perspectives
À partir de ce projet pilote, des démarches ont été entreprises pour établir un consortium international, qui sont toujours en cours. Un bilan des premiers échanges est présenté dans cette section.
4.1 Prospection de nouveaux partenaires
Plusieurs partenaires internationaux potentiels, publics ou privés, ont déjà été approchés. Il s’agit de :
- l’Université de Wageningen aux Pays-Bas (WU) et de l’entreprise Hendrix Genetics, qui disposent déjà d’un total d’au moins 250 séquences individuelles ayant une profondeur au moins égale à 10X ; l’entreprise Hendrix Genetics a financé l’acquisition de la plus grande partie des données proposées et s’est montrée ouverte à l’approche 1000 génomes pour améliorer la connaissance du génome et de la diversité génétique, mais elle ne souhaitait pas que les données soient mutualisées pour la recherche de mutations causales déterminant les QTL ;
- le Friedrich Loeffler Institute (FLI) avec son laboratoire de Mariensee, et l'Université de Göttingen (UGOE) en Allemagne, avec 50 séquences individuelles de profondeur égale ou supérieure à 10X; cette équipe a déjà publié une analyse d'un jeu de 127 séquences obtenues sur des lignées commerciales et l'espèce sauvage Gallus gallus gallus, avec le principal objectif d'identifier des signatures de sélection ou de domestication par la comparaison des fréquences alléliques entre groupes contrastés (Qanbari et al., 2019). Les 2 régions qui apparaissent le plus soumises à sélection sont centrées soit autour du gène ALX1, impliqué dans la morphologie du bec, soit autour du gène KITLG, impliqué dans la pigmentation dans de nombreuses espèces mais pas encore chez la poule.
Les 3 institutions WU, FLI et UGOE ont été partenaires du projet européen IMAGE (http://www.imageh2020.eu/), coordonné par INRAE, qui a financé le séquençage de 300 individus (profondeur 10X) représentant principalement des races locales de poules des 3 pays, France, Allemagne et Pays-Bas. L’intégration des données de ces institutions, des données du projet IMAGE et de celles du projet pilote français porterait le total de séquences individuelles dépassant le seuil de profondeur de 10X de 203 à 803, auxquelles s’ajoutent 72 jeux de données publiques.
Le Roslin Institute détient une centaine de données de séquence et a manifesté un intérêt de principe. Récemment, ce groupe a publié une analyse de la variation des insertions rétrovirales endogènes à partir de 65 données de séquence de génome entier, la plupart provenant de séquences réalisées en mélanges d'ADN (39 mélanges sur 65 données) dont la profondeur moyenne est supérieure à 10X (Mason et al., 2020). Leur étude présente aussi la mise au point d'un pipeline bioinformatique spécifique pour la détection des insertions rétrovirales, qu'il serait intéressant d'appliquer aux données du projet français. Une collaboration est donc souhaitable.
Au niveau international, les autres institutions les plus intéressées sont en Chine et en Corée. Les équipes américaines contactées n'ont pas souhaité s'engager ou bien ont proposé de commencer par stocker les données rassemblées, ce qui n'était pas notre demande. Récemment une étude chinoise a rassemblé 863 séquences pour analyser la diversité des races et la domestication de la poule (Wang et al., 2020). Le coordinateur chinois a produit 787 séquences individuelles, avec une profondeur de 20X pour au moins un individu de chaque espèce ou sous-espèce sauvage, avec de nombreux collaborateurs fournisseurs d'échantillons. La stratégie choisie ici est de rassembler des échantillons pour les séquencer par un seul partenaire, une stratégie complètement différente de celle du projet 1000 génomes. Les résultats sur le scénario de domestication méritent d'être confrontés à d'autres études qui diffèrent par l'échantillonnage des individus sauvages, et un partage de données serait certainement fructueux. Dans cette étude, les résultats sur les signatures de sélection sont commentés pour quelques gènes particuliers impliqués dans le développement (FGFR1, récepteur du facteur de croissance des fibroblastes), ou dans la fonction de reproduction (GNRH-1, hormone contrôlant la libération des gonadotropines, KIF18A, kynésine 18A impliquée dans la mitose des spermatogonies), qui ne sont pas les mêmes que ceux mis en avant par Qanbari et al. (2019). Enfin, l'histoire de la mutation Gly558Arg du récepteur de la thyréostimuline (gène TSHR), absente chez l'ancêtre sauvage et proposée comme une mutation de domestication en 2010, puis confirmée en 2014 comme une mutation favorisée par la sélection de la poule domestique au cours des siècles, apparait assez différente, car la mutation a été identifiée chez une sous-espèce sauvage pour la première fois. L'importance de l'échantillonnage des individus sauvages doit être soulignée, car des phénomènes de croisement incontrôlé avec des poules domestiques pourraient s'être produits.
4.2. Gouvernance et modèle économique
Il paraît donc évident qu’il existe un fort intérêt à regrouper des données de différents projets, mais les modalités de partage doivent être précisées dès lors qu’on sort du contexte français.
Comme pour le « Mille Génomes bovin », un accord de consortium est nécessaire pour organiser la coopération entre partenaires internationaux. Dans le cas du projet bovin, l’accord de consortium a été signé par toutes les institutions apportant des données mais le comité de pilotage est restreint aux membres fondateurs qui ont constitué le consortium initial et apporté les données du premier «run». L’objectif de l’accord est de définir les droits et devoirs des partenaires, de fixer les conditions d’entrée, d’organiser la coordination des études afin de définir celles qui sont réalisées en commun et celles qui peuvent être réalisées en interne par chaque partenaire, et de préciser les conditions de valorisation des résultats. En particulier, toutes les études doivent être connues du comité de pilotage, doivent citer l’origine des données et, en cas de propriété intellectuelle, une licence gratuite est attribuée à tous les membres et leurs partenaires ayant fourni des données, ce qui constitue aussi une forte motivation pour intégrer le consortium. Le consortium bovin est limité à des organismes académiques mais la plupart sont associés à des sélectionneurs privés ou coopératifs dont les intérêts sont pris en compte car ils possèdent les animaux séquencés et ont parfois contribué au financement des recherches. Des sélectionneurs concurrents entre eux ont donc accepté de mutualiser leurs données de séquence afin d’en extraire plus d’informations pour leur usage interne.
Le projet pilote français implique des équipes (Tableau 1) qui se réunissent régulièrement dans le cadre du réseau de génétique avicole d’INRAE, actuellement coordonné par l’unité BOA à Tours. Seules les données du projet UtOpIGe ont été obtenues sur une lignée commerciale de l’entreprise NOVOGEN dans le cadre d’un projet financé par l’ANR qui a fait l’objet d’un accord de consortium. Les conditions d’utilisation de ces données sont donc déjà définies par cet accord initial qui doit continuer à être respecté pour la participation au projet « Mille Génomes ». Il en sera de même pour tous les projets apporteurs de données sur d’autres populations pour lesquels un accord contractuel aura déjà été établi.
Les conditions d’entrée actuellement proposées pour constituer un consortium « Mille Génomes Gallus » sont les suivantes :
i) données de séquence individuelles ayant une profondeur minimale de 10X ;
ii) apport minimum de 300X (30 animaux à 10X), considérant que le seuil initial de 100X choisi pour le projet bovin correspond environ à 300 Gb et donc à 300 génomes de poule ;
iii) le type de populations séquencé est aussi un paramètre à considérer afin de pouvoir disposer d’un nombre significatif d’individus par catégorie de populations, mais ce paramètre ne semble pas limitant car les données de la littérature montrent une grande variété de populations séquencées.
Les données de séquençage obtenues sur mélanges d’ADN sont acceptées en supplément, elles sont plus difficiles à mutualiser car elles sont soumises à un aléa supplémentaire, avec une profondeur moyenne par génome généralement faible et potentiellement hétérogène entre les individus d’un mélange. Le choix du séquençage sur mélange est généralement fait pour limiter les coûts et la profondeur totale obtenue sur un mélange est le plus souvent moyenne.
Le modèle économique initialement envisagé était calqué sur le projet bovin : les membres financent l’acquisition des données qu’ils apportent et le coordinateur finance le stockage des données et la constitution du fichier global des variants, avec sa mise à jour en cas d’évolution de la séquence de référence. Le cout de stockage annuel de 500 séquences (10X) varie de 6000 à 12000 euros selon que les données restent immédiatement accessibles (stockage ‘chaud’) ou non (stockage ‘froid’), ce qui représente chaque année moins de 10% du cout total d’obtention de 500 séquences 10X, estimé à 150 000 euros. Le coût le plus important est celui des ressources humaines en charge des analyses.
Toutefois, la prise en charge du stockage de l'ensemble des données n'a pas été acceptée par INRAE à l'heure actuelle et la capacité à stocker sur un même serveur l'ensemble des données afin de permettre une analyse globale apparait actuellement comme le point bloquant. Les partenaires interrogés en octobre 2019 n'ont pas pris position sur une contribution au coût de stockage et le partenaire chinois n'a pas proposé de rassembler l'ensemble des séquences, considérant sans doute qu'il en avait assez en propre. Pourtant, l'expérience du 1 000 génomes bovins montre l'intérêt d'aller au-delà du millier de séquences, à condition qu'il y ait un accord clair sur la stratégie d'analyse et de partage des résultats. Cet accord reste à construire avec une communauté scientifique et ses partenaires privés qui sont sans doute plus individualistes que ne le sont ceux travaillant sur la génétique bovine. La force de frappe en analyse de données doit aussi être adaptée au volume de données. Il ne s'agit pas seulement de capacité de calcul mais aussi de choix de méthodes, qui vont de l'analyse simple de fréquences alléliques à des méthodes de maximum de vraisemblance plus complexes. L'utilisation de deux méthodes différentes permet d'augmenter la fiabilité des résultats, comme cela a été illustré chez la pintade sur des données de séquences en mélange (Vignal et al., 2019).
Conclusion
La cible de 1000 séquences du génome de la poule en profondeur 10X est atteinte au niveau international mais le regroupement des données et la standardisation de leur description, en fonction des populations concernées et des critères de qualité de séquence, ne sont pas atteints. L’exemple du modèle bovin a montré une dynamique d’attractivité croissante du projet, encouragée par les premiers résultats obtenus sur l’identification d’anomalies génétiques dans les populations commerciales, grâce à la combinaison des données du projet 1000 Génomes et des génotypes disponibles en routine. Dans le cas du projet pilote français, une telle analyse n’est pas réalisable actuellement par insuffisance d’animaux génotypés dans les populations représentées par des animaux séquencés. En revanche, trois types d’analyse sont dès maintenant réalisables :
i) améliorer notre connaissance du processus de domestication et de l’évolution des espèces du genre Gallus ;
ii) identifier les régions du génome et les mutations causales de phénotypes discriminant des populations de poules domestiques ;
iii) produire un inventaire des variants structuraux dans le génome de la poule, qui n’a jamais encore été réalisé sur un effectif aussi important représentatif des principaux types de populations, sauvages et domestiques.
La première action à engager serait la signature d’un accord de consortium avec les différentes institutions intéressées afin de définir les règles de partage et de valorisation des données de séquence, mais la communauté n’y semble pas encore prête. La solution est donc d’élargir le champ d’action du projet pilote français à une collaboration européenne, dans la dynamique lancée par le projet européen IMAGE, en recherchant un financement complémentaire dans les appels à projets nationaux, bilatéraux (notamment le programme ANR-DFG franco-allemand) ou européens. Dans le même temps, des études ciblées vont être réalisées au sein du projet français. Un délai maximal de 2 ans doit permettre de valoriser les données du projet français et de concrétiser, ou pas, une extension européenne.
Remerciements
Les auteurs remercient les porteurs de projets nommés dans le tableau 1 qui ont apporté leurs données à ce projet pilote.
Notes
- Pour plus d’information sur ce protocole consulter le portail de l’infrastructure RARe : https://agrobrc-rare.org
Références
- Alexander D., Novembre J., Lange K., 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res., 19, 1655-1664.
- Bouwman A.C., Daetwyler H.D., Chamberlain A.J., Ponce C.H., Sargolzaei M., Schenkel F.S., Sahana G., Govignon-Gion A., Boitard S., Dolezal M., Pausch H., Brøndum R.F., Bowman P.J., Thomsen B., Guldbrandtsen B., Lund M.S., Servin B., Garrick D.J., Reecy J., Vilkki J., Bagnato A., Wang M., Hoff J.L., Schnabel R.D., Taylor J.F., Vinkhuyzen A.A.E., Panitz F., Bendixen C., Holm L.E., Gredler B., Hozé C., Boussaha M., Sanchez M.P., Rocha D., Capitan A., Tribout T., Barbat A., Croiseau P., Drögemüller C., Jagannathan V., Jagt C.V., Crowley J.J., Bieber A., Purfield D.C., Berry D.P., Emmerling R., Götz K.U., Frischknecht M., Russ I., Sölkner J., Van Tassell C.P., Fries R., Stothard P., Veerkamp R.F., Boichard D., Goddard M.E., Hayes B.J., 2018. Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nature Genet., 50, 362-367.
- Daetwyler H.D., Capitan A., Pausch H., Stothard P., van Binsbergen R., Brøndum R.F., Liao X., Djari A., Rodriguez S.C., Grohs C., Esquerré D., Bouchez O., Rossignol M.N., Klopp C., Rocha D., Fritz S., Eggen A., Bowman P.J., Coote D., Chamberlain A.J., Anderson C., VanTassell C.P., Hulsegge I., Goddard M.E., Guldbrandtsen B., Lund M.S., Veerkamp R.F., Boichard D.A., Fries R., Hayes B.J., 2014. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nature Genet., 46, 858-865.
- Hayes B.J., Daetwyler H.D., 2019. 1000 Bull Genomes Project to Map Simple and Complex Genetic Traits in Cattle: Applications and Outcomes. Ann. Rev. Anim. Biosci., 7, 89-102.
- Li G., Chen S., Duan Z., Qu L., Xu G., Yang N., 2013. Comparison of protoporphyrin IX content and related gene expression in the tissues of chickens laying brown-shelled eggs. Poult Sci., 92, 3120-3124.
- Ling M.K., Lagerström M.C., Fredriksson R., Okimoto R., Mundy N.I., Takeuchi S., Schiöth H.B., 2003. Association of feather colour with constitutively active melanocortin 1 receptors in chicken. Eur. J. Biochem., 270, 1441-1449.
- Mason A.S., Lund 1.R., Hocking P.M., Fulton J.E., Burt D.W., 2020. Identification and characterisation of endogenous Avian Leukosis Virus subgroup E (ALVE) insertions in chicken whole genome sequencing data. Mobile DNA, 11, 22.
- Qanbari S., Rubin C. J., Maqbool K., Weigend S., Weigend A., Geibel J., Kerje S., Wurmser C., Townsend Peterson A., Brisbin Jr. I.L., Preisinger R., Fries R., Simianer H., Andersson L., 2019. Genetics of adaptation in modern chicken. PLoS Genet., 15, e10077989.
- Romé, H., Varenne A., Herault F, Chapuis H., Alleno C., Dehais P., Vignal A., Burlot T., Le Roy P., 2015. GWAS analyses reveal QTL in egg layers that differ in response to diet differences. Genet. Sel. Evol., 47, 83.
- Samiullah S., Roberts J.R., Chousalkar K., 2015. Eggshell color in brown-egg laying hens - a review. Poult. Sci., 94, 2566-2575.
- Vignal A., Besbes B., 2005. Séquençage du génome de la poule et perspectives d'application en poule pondeuse. Journ. Rec. Avicole, St Malo, France, 6, 506-513.
- Vignal, A., Boitard, S., Thebault, N., Dayo G. K., Yapi-Gnaore, V., Isaaka Y., Berthouly-Salazar C., Palinka-Bodzsar N., Guemene D., Thibaud-Nissen F., Warren W.C., Tixier-Boichard M., Rognon X., 2019. A guinea fowl genome assembly provides new evidence on evolution following domestication and selection in galliformes. Mol. Ecol. Resour., 19, 997-1014
- Wang, M., Thakur, M., Peng, M., Jiang Y., Frantz L.A.F., Li M., Zhang J.J., Wang S., Peters J., Otecko N.O., Suwannapoom C., Guo X., Zheng Z.Q., Esmailizadeh A., Hirimuthugoda N.Y., Ashari H., Suladari S., Zein M.S.A., Kusza S., Sohrabi S., Koopaee H.K., Shen Q.K, Zeng L., Yang M.M., Wu Y.J., Yang X.Y., Lu X.M., Jia X.Z., Nie Q.H., Lamont S.J., Lasagna E., Ceccobelli S., Gunwardana H.G.T.N., Senasige T.M., Feng S.H., Si J.F., Zhang H., Jin J.Q., Li M.L., Liu Y.H., Chen H.M., Ma C., Dai S.S., Bhuiyan A.K.F.H., Khan M.S., Silva G.L.L.P., Le T.T., Mwai O.A., Ibrahim M.N.M., Supple %M., Shapiro B., Hanotte O., Zhang G., Larson G., Han J.L., Wu D.D., Zhang Y.P., 2020. 863 genomes reveal the origin and domestication of chicken. Cell Res., 0, 1-9.
Résumé
Le séquençage du génome entier est devenu abordable chez la poule. Le projet « Mille Génomes Gallus » a pour objectif de rassembler les séquences d’animaux du genre Gallus, produites dans des projets de recherche. L’intérêt est d’augmenter la puissance des analyses génomiques en augmentant le nombre de génomes comparés. Les applications possibles concernent l’analyse de la structure du génome, la caractérisation globale de la diversité de l’espèce, l’identification de mutations causales et l’aide à la sélection génomique. Cette synthèse décrit d’abord le contexte de la génomique de la poule avant de développer le concept de « Mille Génomes », de l’illustrer avec un projet pilote porté par des équipes françaises et de discuter les modalités de son extension. Le projet pilote français regroupe 207 séquences individuelles pour 8 projets de recherches financés par des fonds publics, sur une large gamme de populations (poulets de chair, poules pondeuses, races locales, espèces sauvages). Les jeux de données sont décrits par des métadonnées techniques et des métadonnées liées à l’animal et sa population d’origine. Aucune information phénotypique n’est partagée. La comparaison des séquences à la version 5 du génome de référence a permis de répertorier plus de 40 millions de variants SNP. L’analyse de structure des populations a identifié sept groupes génétiques. Le gène MC1R a été choisi comme exemple permettant de détecter une signature de sélection chez les pondeuses de plumage rouge et d’autres gènes candidats sont en cours d’étude sur la qualité de coquille. D’autres données produites en Europe et en Chine montrent que 1000 génomes de poule ont déjà été séquencés. Les principes d’un accord de consortium sont exposés afin d’élargir notre projet à un plus grand nombre de données de séquence.
Pièces jointes
Pas de document complémentaire pour cet articleStatistiques de l'article
 Vues: 2626
Vues: 2626
Téléchargements
 XML: 296
XML: 296
 PDF: 603
PDF: 603
Articles les plus lus par le même auteur ou la même autrice
- Alicia JACQUES, Delphine DUCLOS, Michèle TIXIER-BOICHARD, Gwendal RESTOUX, Les ressources génétiques cryoconservées pour restaurer et conserver la diversité des animaux d’élevage , INRAE Productions Animales: Vol. 38 No 3 (2025)