

Epigenetics and the elaboration of phenotype in cattle (Full text available in English)
Header
Because of their important role in human health, epigenetic marks are the subject of increasing attention in both the scientific literature and the media. But what about the animal world, and particularly farmed livestock? This review takes stock of the knowledge acquired at INRAE concerning epigenetic mechanisms in cattle and prospects for their future use in livestock farming.
Introduction
In animal production, and particularly in cattle, the objective is to increase productivity while respecting the health and well-being of the animals. Major efforts regarding genetic selection have been made during the past fifty years and many genetic markers have been associated with milk production, meat quality, reproduction and growth traits (Bouquet and Juga, 2013; Boichard et al., 2016). However, genetics can only explain part of the phenotypic variability of traits of interest to breeders; epigenetic modifications also contribute to this variability.
Originally used to describe the influence of the environment on the development of phenotypes, the term "epigenetics" has evolved considerably since it was introduced in 1942 by Conrad H. Waddington. Epigenetics is now defined as the set of marks on the genome that induce changes in gene expression without altering the DNA sequence (Berger et al., 2009). These marks are both stable and heritable during cell division. The epigenome of a cell is its complete set of epigenetic marks, including DNA methylation, post-translational histone changes, chromatin remodelling, non-coding RNAs and other molecules that can transmit information through mitosis by regulating gene expression.
The epigenome is highly dynamic throughout life, and is governed by complex interactions between genetic and environmental factors (Kouzarides, 2007). Indeed, all cells in an individual possess the same genetic heritage that they use in different ways, expressing different genes to a greater or lesser extent depending on the physiological stage and cell type. This information, which is specific to a given cell state, is orchestrated by activating or inhibitory epigenetic marks that can modify gene expression and define specific phenotypes. Moreover, epigenetic marks are modifiable and/or reversible depending on the environment, and these modifications can also have long-term consequences (Figure 1). The development of an individual (embryo, foetus and newborn, and also the differentiation and maturation of germ cells) and more generally the period surrounding conception, represent a window that is particularly sensitive to different environmental factors. Indeed, the return to totipotency necessary to initiate the developmental programme involves considerable plasticity of the epigenetic marks in terms of their diversity, quantity and duration of action. Environmental variations that occur during gestation can therefore exert effects on the phenotype of the next generation, and even beyond (Junien et al., 2016).
In this article, we briefly describe these epigenetic processes, and then use several examples that mainly result from work carried out at INRAE to discuss the crucial role of the epigenome in constructing the phenotype in cattle, with particular emphasis on fertility, developmental, health and milk production traits.
Figure 1: Contribution of the epigenome to phenotype variability.
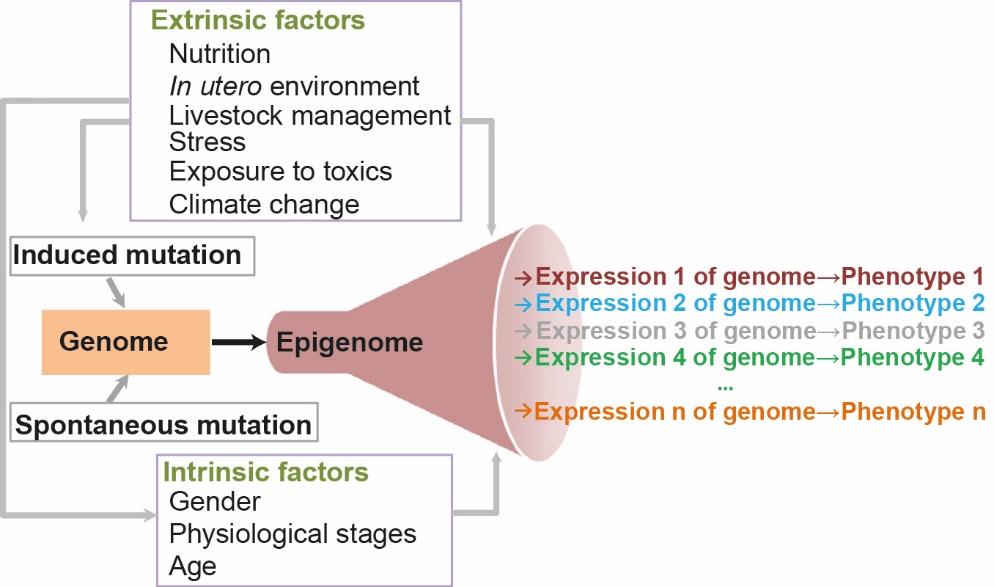
The genome evolves over time under the influence of randomly occurring mutations; these are maintained (or not) within populations depending on their functional repercussions. Epigenetics enables the specific reading of genetic information that contributes to the establishment of differential transcriptional states within each cell type in each tissue. The epigenome is also modified by extrinsic and intrinsic factors, enabling a plasticity of genome expression and a declination of phenotypic responses.
1. Epigenetic marks and mechanisms
1.1 DNA methylation
Within the DNA sequence, the cytosine residues of CpG dinucleotides (cytosine-phosphate-guanine) can be methylated to 5-methylcytosine (5meC; Figure 2). This DNA methylation has been known for more than 50 years, following the discovery of 5meC in the sea urchin embryo and then linking this epigenetic mark with the activity of genes during development. It is present in almost all living organisms, but in mammals only 5% to 10% of cytosines in the genome are methylated. This percentage seems low, but compared to CpG dinucleotides it often exceeds 80%.
CpG sites are not distributed evenly along the genome. The regions with the highest density of CpG sites are called "CpG islands" and are generally methylated when associated with repetitive elements of the genome, such as retrotransposons (mobile elements of the genome of viral origin that have accumulated during evolution) and centromeric and pericentromeric satellite sequences. DNA methylation at the level of retrotransposons is necessary to prevent their replication and protect the genome against their invasion. The methylation of pericentromeric satellite sequences is involved in the formation of constitutive heterochromatin, which is essential to limit undesirable chromosomal recombination and segregation. In regions flanking the CpG islands, DNA methylation is particularly dynamic as a function of cell type, developmental stage, the animal’s physiology or the environment.
Associated with regulatory elements or promoters, DNA methylation inhibits gene expression, while at the intragenic level it has an activating role by limiting the initiation of illegitimate transcription (Schubeler, 2015). DNA methylation also intervenes in X chromosome inactivation, which can compensate for the double allele burden in females, as well as in the genomic imprinting phenomenon described for around 100 genes in humans and mice. Genes subject to imprinting express in a mono-allelic manner depending on the parental origin of the allele; this mono-allelic expression is essential for a satisfactory course of development and harmonious growth of the foetus. These genes do not appear to be completely conserved between species, and their list in cattle is not yet exhaustive.
The functions of DNA methylation are mediated by nuclear proteins carrying a methylated DNA binding domain and able to recruit transcriptional repressors or histone modifying enzymes. DNMTs (DNA methyltransferases) are the enzymes that catalyse the transfer of methyl groups to the 5-position of cytosines from the S-adenosylmethionine metabolite (produced from dietary folic acid). The DNMT3A and DNMT3B enzymes are involved in the de novo methylation that takes place during cell differentiation processes, but also in response to a stimulus in differentiated cells. The DNMT1 enzyme, which recognises the hemimethylated CpG sites resulting from DNA replication, ensures the maintenance and propagation of methylation patterns through cell division (Lyko, 2018). Demethylation may thus result from a lack of DNMT1 activity, as DNA methylation is progressively diluted during cell cycles. Active DNA demethylation mechanisms are also observed during the waves of epigenetic reprogramming that occur in germ cell precursors and the preimplantation embryo. These mechanisms involve the conversion of 5meCs by TET enzymes into oxidised derivatives such as 5-hydroxymethylcytosine (5hmC), which are then diluted during replication or replaced by unmethylated cytosines by the DNA repair machinery (Wu and Zhang, 2017).
Figure 2: Epigenetic regulation of gene expression.
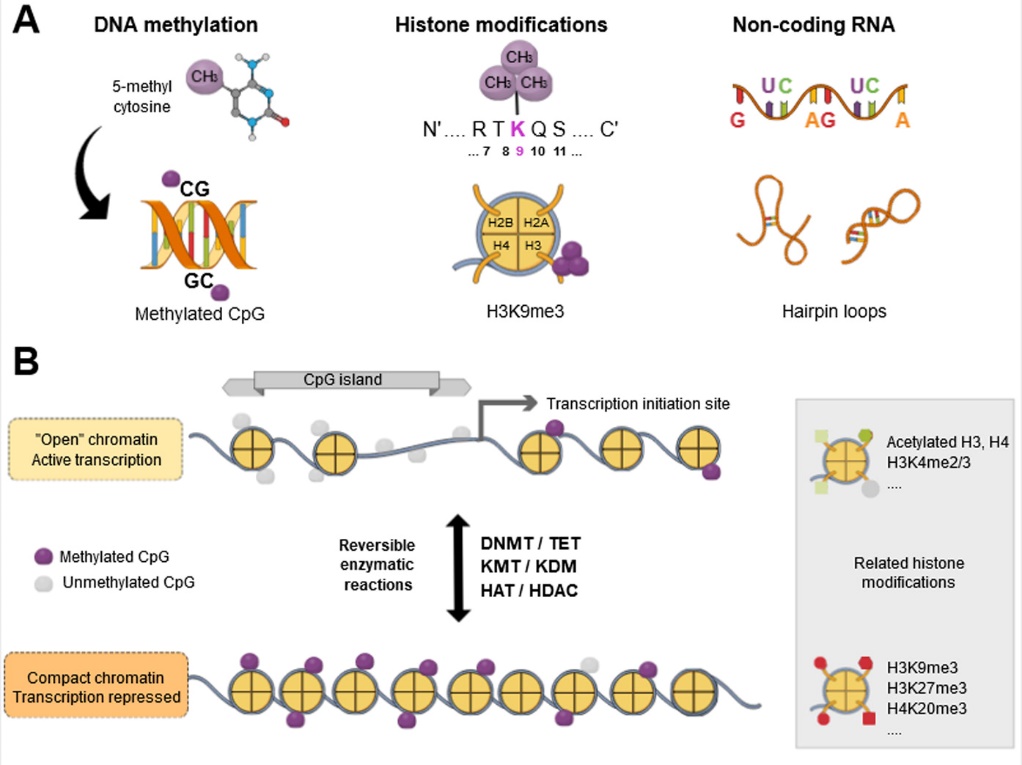
A. DNA methylation on the cytosines of CpG dinucleotides, histone modifications in nucleosome cores and non-coding RNAs that sometimes adopt specific, so-called hairpin-loop structures are the main epigenetic regulators presented here. In the case of histone modifications, mention should be made of the target histone, the residue and its position relative to the N-terminal tail, and then the type and number of modifications (e.g. H3K9me3 for the trimethylation of histone H3 on lysine at position 9).
B. Depending on the environment, different epigenetic marks may be present, particularly at the level of the CpG islands upstream of the gene transcription initiation site, conferring a more or less "open" chromatin structure. Epigenetic modifications are reversible. This process is mediated by several families of enzymes with antagonistic activities: DNMT and TET for DNA methylation, KMT and KDM for histone methylation, and HAT and HDAC for histone acetylation. Figure produced on www.mindthegraph.com
1.2 Post-translational histone modifications
In the nucleus, genomic DNA is wrapped around octamers of four types of histones (H2A, H2B, H3 and H4) to form the nucleosome (Figure 2). The histones are small basic proteins, which facilitates their binding to DNA, and they also contain N-terminal tails targeted by different types of modifications: acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ribosylation, deamination and isomerisation. These modifications affect different amino acids, producing dozens of post-translational variants with different functional roles. The addition or removal of these modifications are highly flexible processes that directly affect the accessibility of genomic DNA to the transcription machinery, and thus the activation or repression of gene expression (Kouzarides, 2007). The combinatorial nature of the different histone marks ("histone code"), together with DNA methylation and the presence of certain transcription factors or RNA polymerase, define specific chromatin states associated with specific transcriptional states. These chromatin states are transmitted to daughter cells, thus ensuring the continuity of cell identity through mitosis.
Histone modifications are based on the use of dietary metabolites (acetyl-coA and S-adenosylmethionine) and are controlled by an extensive epigenetic machinery that includes enzymes capable of affixing, erasing and reading these marks. Acetylation and deacetylation were described as early as the 1960s (Allfrey and Mirsky, 1964). Resulting from the activity of histone acetyltransferases (HATs), acetylations neutralise the positive charge of the histone, increase its steric hindrance and decrease the strength of its interaction with DNA. Acetylated lysines also permit the recruitment of bromodomain proteins that are involved in chromatin remodelling. These different modes of action result in opening of the chromatin (euchromatin) and a local increase in transcriptional activity (Mujtaba et al., 2007). Deacetylation processes, which exert an opposite action on transcription, involve histone deacetyltransferases (HDACs) and are associated with the formation of heterochromatin (compact chromatin). Methylations, which consist in the addition of one or more methyl group(s) to the lysine or arginine residues of the histone tails, are catalysed by histone methyltransferases (KMT), while demethylations result from the activity of histone demethylases (KDM) (Jenuwein, 2006). At the H3K4 position, trimethylation (H3K4me3) is a signature of transcriptional activity, particularly when combined with acetylation. Conversely, methylation is associated with a repression of transcription when it affects H3K27 and H4K20 and with the constitutive heterochromatin at position H3K9, in association with the HP1 protein and DNA methylation (Nishibuchi and Dejardin, 2017).
Finally, it is important to note that one cell type escapes from this nucleosome structure: the spermatozoon. Depending on the species, 85 to 99% of the histones are replaced by protamines, arginine-rich proteins that form toroid-shaped structures with DNA (Carrell, 2012). The replacement of histones by protamines plays an important role in sperm chromatin compaction, which contributes to reducing nuclear volume and the acquisition of a hydrodynamic morphology, and makes it possible to protect the paternal genetic heritage against oxidation, particularly during migration through the epididymis and female genital tract.
1.3. Non-coding RNAs
The discovery of non-coding RNAs overturned the dogma that each gene coded for a protein with a cellular function. Recent research has highlighted many forms of non-coding RNAs, highlighting their roles in physiology and their involvement in numerous pathologies (Bayoumi et al., 2016). Non-coding RNAs are divided into subclasses according to their size, function or genomic location. Small non-coding RNAs are those smaller than 200 nucleotides, and include microRNAs (miRNA, 19-24 nucleotides). In mammals, more than 2,000 miRNAs per species are currently listed in the public miRBase reference database. At the genomic level, miRNAs can be organised in clusters (Griffiths-Jones et al., 2008); they then share a common promoter and are transcribed into large polycistrons (large RNA fragments containing several miRNAs). The genes encoding miRNAs can also be located in the introns of coding or non-coding genes. These intronic miRNA genes may either share the same promoter as their host or use a separate promoter or even several sites of transcription initiation (Monteys et al., 2010).
The biosynthesis of functional miRNAs and assembly of the RISC complex ("RNA-induced silencing complex") involves a five-stage cascade of enzymatic reactions: i) transcription of the miRNA-encoding gene into primary miRNA (pri-miRNA), ii) maturation of the pri-miRNA into a precursor (pre-miRNA) at the nuclear level, iii) export of the pre-miRNA from the nucleus to the cytoplasm, iv) maturation of the pre-miRNA into mature miRNA and v) formation of the RISC complex. This complex is guided by the miRNA to its target transcripts by partial sequence homology between the two RNA species; it can then induce the inhibition of translation or the degradation of messenger RNAs. A miRNA can thus target a hundred or so different mRNAs and an mRNA can be targeted by several dozen different miRNAs. By participating in the regulation of gene expression, miRNAs play key roles in all biological functions in mammals (Bushati and Cohen, 2007). The functions of miRNAs in proliferation, differentiation or cell death have been conserved during evolution and come into play in all biological pathways, such as the immune response, circadian rhythm or brain development.
Studies of miRNA expression profiles have indicated that the majority of miRNAs are controlled by developmental and/or tissue-specific signals, such as miR-1 which accounts for 45% of miRNAs expressed in the heart and miR-122 which accounts for 72% of miRNAs in the liver (Lagos-Quintana et al., 2002). Precise control of miRNA expression levels is crucial for maintaining the physiological functions of the cell, and the deregulation of miRNA expression is often associated with diseases such as cancer (Landgraf et al., 2007). Their deregulation may play a role in tumour cell initiation, proliferation and invasion processes; recent studies also seem to implicate them in the mechanisms of resistance to certain anti-tumour treatments. The deregulation of miRNA expression in cancer may also involve epigenetic mechanisms that affect the activity of their promoters (Choudhry and Catto, 2011).
MiRNAs can be secreted into the extracellular medium and circulate in biological fluids (Weber et al., 2010). In many pathological situations, quantitative or even qualitative variations in circulating miRNAs in serum (of peripheral origin), platelets, leukocytes and red blood cells have been highlighted, so they may also constitute potential biomarkers.
2. Fertility, reproduction and development
2.1. Epigenetic reprogramming in germ cells
Germ cell (GC) differentiation takes place in the male (testis) and female (ovaries) gonads. During foetal life, the gonads first appear as genital ridges and then differentiate into an ovary or testis depending on the sex of the individual. This male or female gonad orientation governs the fate of the germline and the nature of associated epigenetic remodelling. The establishment of male and female germlines requires three successive stages: i) specification of primordial GCs from the embryonic epiblast at the start of gastrulation, ii) migration towards the genital ridges, and iii) differentiation according to the testicular or ovarian environment. While male GCs stop proliferating and enter quiescence (meiosis starting at puberty), female GCs enter meiosis during foetal life. These three stages are based on a sex-specific transcriptional programme orchestrated by an extensive epigenetic reprogramming. The DNA methylation pattern characterising the embryonic epiblast is thus erased throughout the genome when the primordial GCs colonise the gonad; DNA methylation is then re-established according to two different dynamics in male and female GCs (Figure 3).
Figure 3: Evolution of DNA methylation in germ cells (GCs) during development.
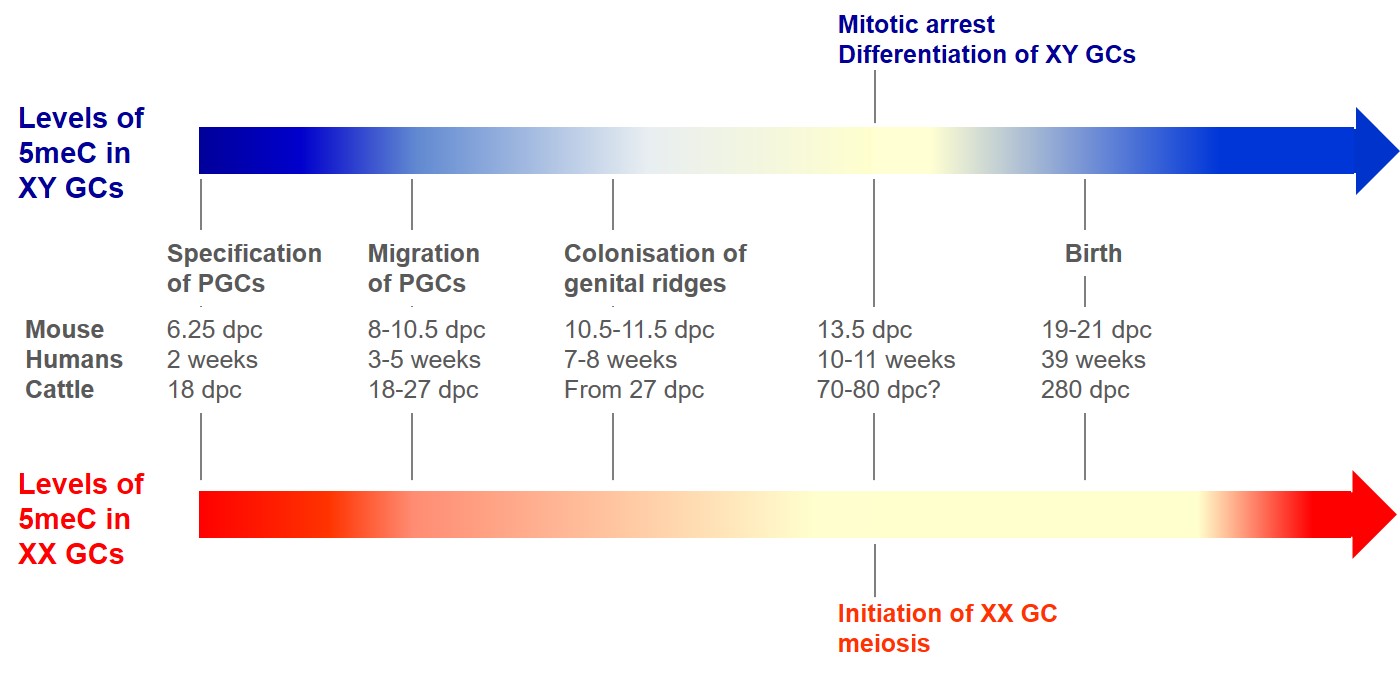
The colour intensity of the arrows indicates the levels of DNA methylation in GCs as a function of developmental stages in mice. Thus, during their migration and then their colonisation of genital ridges, GCs gradually demethylate their genome, to reach the lowest level of methylation at the stage preceding meiosis in the ovary and the cessation of GC mitosis in the testis. In males a new methylation pattern is rapidly established, whereas in females, DNA re-methylation in the oocyte will start during the oocyte maturation of growing follicles. The evolution of DNA methylation in bovine foetal GCs is not known; the equivalence of developmental stages is given as an indication. dpc: day post coïtum; PGC: primordial germ cell.
Most knowledge concerning the dynamics of the erasure of DNA methylation is based on data from the mouse model and, to a lesser extent, on information acquired in humans. DNA methylation erasure first affects most of the CpG islands in the genome of migrating primordial GCs, and then more specific regions during the colonisation of genital ridges. This erasure is not total: some genomic regions, such as repetitive elements, retain methylation to a degree that differs as a function of sex and species (Seisenberger et al., 2012; Guo et al., 2015). In parallel with DNA demethylation, the levels of repressive histone marks rise in order to prevent the initiation of massive transcriptional activity following genome hypomethylation (Gkountela et al., 2015). The loss of 5meC and then of repressive histone marks, as well as the gain in 5hmC, are essential for the expression of germline differentiation genes and the progression of gametogenesis, particularly in females where it just precedes the initiation of meiosis (Hill et al., 2018).
The DNA re-methylation of GCs uses the de novo methylation enzymes DNMT3A and 3B, and also DNMT3L, a germline specific cofactor, which is devoid of methyl transferase activity and guides DNMT3A and B to the sequences to be methylated. This step occurs at different stages according to sex. In males, de novo methylation begins as early as foetal life. Although most methylation in male GCs is acquired during life in utero, it appears that modifications still occur after birth and even during spermatogenesis (Ly et al., 2015). The dynamics of de novo methylation is not yet understood in male cattle, but is under study at INRAE (Mandon-Pépin et al., 2017). In females, de novo methylation occurs progressively after puberty as the oocyte matures and grows larger during follicular growth. The first signs of re-methylation appear at the primary to secondary follicle transition when the oocyte has reached 110µm in the cow (O'Doherty et al., 2012). De novo methylation then continues until ovulation.
Thus the de novo methylation of GC DNA occurs mainly during foetal life in males and after puberty in females. Male and female GCs therefore do not display the same sensitivity to the environment, particularly during gestation. The living conditions of the mother can have an impact on the reprogramming of male GCs and the sperm epigenome in adulthood, with physiological consequences, particularly in terms of metabolism, that can be seen in the next generation (Radford et al., 2014). In cattle, environmental control during gestation (and particularly the diet of highly producing dams) may therefore prove crucial to ensuring future bull fertility.
2.2. Sperm epigenome
a. DNA methylation
We showed that bovine spermatozoa have a particularly low global level of 5meC compared to bovine somatic cells and also to spermatozoa from goats, rams, humans, stallions, boars and mice (Perrier et al., 2018). This low methylation has been observed in all cattle breeds studied to date and does not seem to be affected by the semen freezing process. To determine the hypomethylated sequences, we compared the sperm methylome with that of bovine somatic cells using pan-genomic approaches. Numerous differentially methylated positions were found, 81% of which were specifically hypomethylated in spermatozoa. These hypomethylated sites were enriched with spermatogenesis genes as well as satellite and ribosomal DNA repeats (Perrier et al., 2018). The over-representation of these repetitive elements in the bovine genome, as well as their low methylation, could explain why bovine spermatozoa have a lower global 5meC level than spermatozoa from other mammalian species. This work, combined with similar studies in other species, has enabled us to propose a typical profile of the sperm methylome (Kiefer and Perrier, 2019; Figure 4).
Factors associated with variations in the sperm methylome are now being studied in a cohort of 260 bulls. Their breed (Holstein, Montbéliarde, Normande, Charolais, Blanc Bleu Belge and Abondance) seems to shape the spermatic methylome (Perrier et al., 2019). An important effect of the bull's age at the time of semen collection was also found in all the breeds studied, which suggests an unexpected plasticity of the spermatic epigenome, which until recently was considered to be stable in adulthood as the wave of de novo re-methylation is then completed. Since 2017, several publications in mice, humans and more recently in cattle (Lambert et al., 2018; Takeda et al., 2019), have supported these observations. This raises questions about the possible consequences of current semen collection practices, which are being carried out at an increasingly early age in dairy breeds. Artificial intelligence approaches are currently being conducted to isolate the best biomarkers predictive of male fertility in the sperm methylome.
Since the advent of genomic selection, the genetic value of sires is established at an early stage based on their genotypes, and numerous methods to accelerate sexual maturity, and therefore the sale of semen, have been tested. One such practice is offering young bulls a high plane of nutrition during the first months of life, which results in triggering puberty one month early with no visible effects on semen quality (Byrne et al., 2018). We have shown that the effects of diet and advanced puberty on sperm DNA methylation are relatively limited, but they do exist (Perrier et al., 2020). These data suggest that the sperm epigenome contains a memory of past events, and that the first months of the bull's life represent a window of sensitivity to environmental variations. It would be important to determine the degree to which these methylation variations might alter the development and phenotype of progeny.
Figure 4: Relationships between functional elements of the genome, CpG density and 5meC in somatic cells and spermatozoa.
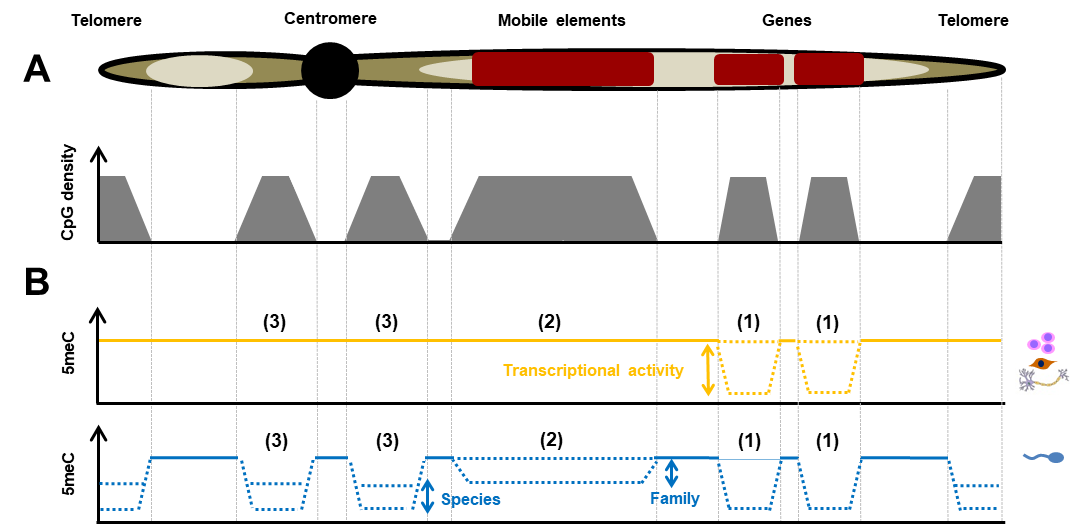
CpG density (A) and DNA methylation (B) in different elements of the genome are plotted along a chromosome. In B, the solid lines illustrate the overall hypermethylation of the genome, which appears to be conserved between mammalian species and differentiated cells, while variable methylation is represented by dotted lines (yellow: somatic cells; blue: spermatozoa). The margins and factors of variation are indicated by arrows. Gene promoters (1), which have variable methylation depending on transcriptional activity in somatic cells, are demethylated in transcriptionally inactive spermatozoa. Mobile elements of the genome (2) and pericentromic sequences (3), which are hypermethylated in somatic cells, display variable methylation depending on the family of retrotransposons that constitute them (2) and the species (3), respectively. According to (Kiefer and Perrier, 2019).
b. Small non-coding RNAs
The sperm RNA content has long been regarded as a remnant of spermatogenesis. Recent studies have shown that spermatic RNAs are transferred into the oocyte and contribute to successful fertilisation and early stages of embryonic development. Small non-coding RNAs (sncRNAs) in particular may be biomarkers that reflect the successful course of spermatogenesis and/or semen functionality (Bissonnette et al., 2009).
An exhaustive analysis of the expression profiles of sperm sncRNAs in a cohort of 40 bulls from six breeds (Holstein, Montbéliarde, Normande, Charolais, Blanc Bleu Belge and Abondance) was recently carried out (Sellem et al., 2020). In addition to miRNAs (20%), we identified RNAs interacting with PIWI (piRNA, 26%) and also fragments of ribosomal RNAs (25%) and transfer RNAs (14%). It is interesting to note that the fragments of transfer RNAs associated with glycine or glutamine were highly represented among all transfer RNAs. Variations in expression profiles were observed between breeds for all categories of sncRNAs.
Only 26% of the sperm miRNAs identified in our data have already been described, the vast majority being predicted and potentially specific to the bovine species. We also showed that 2% of the miRNAs were very strongly expressed in spermatozoa, suggesting their functional importance. Studies have indicated a potential correlation between the expression profile of miRNAs and bull fertility (Govindaraju et al., 2012; Fagerlind et al., 2015). The presence of these miRNAs may favour a decline of oocyte messenger RNAs which correspond to the target genes for these miRNAs, and a change to the early stages of embryonic development. Monitoring the effects of the over-expression or suppression of these miRNAs on the kinetics and quality of embryonic development might enable exploration of their function.
2.3 Epigenome of the embryo
After fertilisation, the paternal and maternal genomes are reprogrammed to allow development of the embryo. This reprogramming is characterised by a series of epigenetic modifications that start just after fertilisation (Ross and Sampaio, 2018), particularly in the paternal genome. The protamines present on paternal DNA are exchanged with maternal histones, which are rapidly methylated in position H3K4 (activating mark). By contrast, the maternal chromatin contains numerous repressive histone modifications. This asymmetry between parental genomes that can be seen after fertilisation eventually fades as embryonic development progresses (Pichugin et al., 2010; Boskovic et al., 2012). It has also been shown that overall 5meC levels fall sharply in embryos during pre-implantation development. At the level of the genomic sequence, this decrease is associated with successive waves of demethylation and de novo methylation which coincide with the principal stages of early development (Jiang et al., 2018). As in mice, demethylation is more rapid and important in the bovine paternal genome and requires the expression of TET enzymes. Furthermore, while the DNMT1 and DNMT3A enzymes are present, it appears that DNMT3B is primarily responsible for controlling 5meC levels in bovine embryos. Genes subject to genomic imprinting are not affected by this wave of demethylation.
These epigenetic changes participate in triggering embryonic genome activation (EGA). Indeed, initially, the genome of the newly fertilised embryo is transcriptionally inactive. Embryo development then depends strictly on the stock of RNA and proteins accumulated in the oocyte. EGA occurs at the 8-cell stage in cattle, or three days after fertilisation, and is characterised by a unique chromatin organisation. Thus, the global levels of repressive histone marks that decrease after fertilisation reach a minimum level at EGA and recover to the blastocyst stage, as the first cell differentiation occurs. For example, the H3K27me3 mark is eliminated during the first embryonic divisions by histone methyltransferase KDM6B (Chung et al., 2017). When the expression of KDM6B is repressed, preventing the decline of H3K27me3, this causes a change to EGA and a reduction in blastocyst rate. Such gene invalidation experiments on enzymes catalysing histone modifications have confirmed their essential role in chromatin remodelling after fertilisation and EGA.
In addition, growing numbers of bovine embryos are being produced after superovulation (hormonal stimulation used to increase the number of oocytes), in vitro maturation (IVM) to obtain oocytes prior to in vitro fertilisation (IVF), and in vitro embryo culture. This production method is not without effects on the epigenetic profile of embryos and individuals in the longer term (Beaujean, 2018). Superovulation appears to affect the establishment of epigenetic marks and the oocyte transcriptome, which may impact the developmental capacity of embryos produced under these conditions. We have also shown that the exposure of bovine oocytes to heat shock resulted in an accumulation of H3K9me3 and excessive heterochromatin formation in embryos obtained by IVF (Camargo et al., 2019), illustrating the sensitivity of oocytes to culture conditions. These anomalies are also correlated with an increase in the apoptosis rate of embryonic cells and a reduction in blastocyst rate.
Gamete and embryo storage are widely used in association with reproductive biotechnologies. Abnormally high levels of certain histone modifications have been observed in vitrified bovine oocytes. In cryopreserved bovine embryos, DNMT3A expression and 5meC levels also appear to be affected (Stinshoff et al., 2011). However, such studies are quite rare in farmed livestock. There has not yet been an assessment of the potential impact of these epigenetic modifications on subsequent development of the embryo and newborn.
Finally, large calf syndrome was the first clear evidence of the negative impact of in vitro culture conditions on the development of ruminant embryos after their transfer to recipients (Heyman, 2005). This syndrome is characterised by a proliferation phenotype and seems to be linked to early conditions for embryo culture (presence of foetal calf serum rich in growth factors and/or the addition of these factors to culture media), which have since been rectified. This phenomenon highlighted the persistence during gestation of defects induced by the culture period, affecting DNA methylation, the expression of imprinted genes and histone acetylation (Beaujean, 2018).
2.4. Epigenome abnormalities and developmental defects in bovine clones
Obtaining cloned animals is based on major epigenetic reprogramming of the somatic nucleus by the oocyte machinery, which allows the expression profile of the differentiated donor cell to be replaced by a profile compatible with embryonic development. Using a fibroblast cell line derived from a single cow, it is possible to obtain several individuals with the same genotype. As nuclear transfer is carried out in oocytes of differing origins, each somatic nucleus finds itself in a different maternal environment. The resulting epigenetic reprogramming may present errors and induce a broad range of effects: the immediate arrest of development or foeto-placental syndrome that limit the success of cloning to 12% of viable and healthy individuals (Heyman, 2005).
a. Embryonic development
Elimination of the epigenetic profile associated with the type of donor cell, and its replacement by the epigenetic profile of the embryo, appear to be incomplete or incorrect in a significant proportion of embryos. A clear correlation has been demonstrated between the effectiveness of the reorganisation of nuclear architecture (particularly the H3K9me3 profile) and the percentage of cloned embryos surviving the first embryonic cleavages (Pichugin et al., 2010). Because these abnormal epigenetic profiles have a negative impact on embryonic and foetal development, different strategies have been used to try to correct epigenetic errors and improve cloning efficiency. For example, the overexpression of AID encoding an enzyme involved in active DNA demethylation reduces the hypermethylation of cloned embryos and significantly improves cleavage and blastocyst rates in cattle (Ao et al., 2016). However, as the resulting blastocysts are generally not transferred to recipients, there is limited evidence of the long-term impact of this type of strategy.
b. Extra-embryonic tissues
The effects of nuclear transfer on the development of extra-embryonic tissues at key stages of gestation were analysed in 124 cloned and control conceptuses (Guillomot et al., 2014). In addition to differences in 5meC levels between tissues and stages, this study showed that cloning caused a significant rise in 5meC levels in certain extra-embryonic compartments. This difference then disappeared during later stages of gestation, suggesting the existence of compensation mechanisms. Levels of 5meC are always lower in pathological foetuses whose development has ceased.
c. Perinatal Liver
Epigenetic abnormalities have been reported in the livers of bovine clones that died during the perinatal period (Kiefer et al., 2016). These abnormalities were correlated with major changes to liver function in these animals, such as a reduced glucose reserve in the liver. This reserve normally builds up shortly before birth and is used by newborns until the first food intake. Epigenetic abnormalities affect genes that are important for metabolism, and lead to a deregulation of the expression of TCF7L2, a major gene for predisposition to diabetes in humans involved in the production of glucose by the liver. It can therefore be hypothesised that incomplete reprogramming of the somatic nucleus alters the ability of cloned foetuses to adapt to post-natal feeding conditions. Beyond its implications for cloning, this study (Kiefer et al., 2016) showed that particular attention needs to be paid to the foetus' liver in order to better prepare it for the metabolic transition represented by birth and thus optimise the calf's chances of survival. A diet that meets the needs of the gestating mother while allowing the development of a favourable foetal hepatic epigenome could be considered as a lever to improve liver function.
3. Health and immunity: epigenetic marks of circulating cells
The health of farmed livestock is crucial to their well-being and realisation of their genetic potential. The concept of 'robust and efficient animals' encompasses the notions of health associated with resistance to pathogens and/or an efficient immune response, adaptation to environmental changes (breeding practices, nutrition, climate) and products of quality and quantity while limiting environmental impacts. Phenotype/genotype analyses enable genomic selection to identify such animals. A complementary approach is to focus on the epigenome of immune cells. Innate and adaptive immune responses are the result of complex cell differentiation processes that involve a succession of gene expression profiles, regulated with considerable spatial and temporal precision by specific transcription factors and epigenetic processes. Widely documented in humans or in rodent models (Alvarez-Errico et al., 2015), this field remains little studied in cattle.
3.1 Haematopoiesis and DNA methylation
Pluripotent haematopoietic progenitor cells in the bone marrow are characterised by a basal state of methylation determining a transcriptomic profile in support of their renewal capacities. The commitment of these cells to myeloid and lymphoid differentiation pathways is conditioned by a loss or increase in methylated loci, respectively. Subsequent waves of differentiation into lymphocytes, granulocytes, monocytes, macrophages, eosinophils and neutrophils are also under epigenetic control (Alvarez-Errico et al., 2015) (Figure 5A). Epigenetic mechanisms control not only cell differentiation, but also the function and activation of each subpopulation, and thus form the basis for plasticity of the immune response. Each immune subpopulation can therefore be characterised by discriminating loci that carry specific methylation signatures.
Different subpopulations of circulating leukocytes have been described in cattle, as well as their variations in proportion to age, physiological stage and exposure to pathogens. Monocyte subpopulations may notably explain variations in responses to infection, inflammation and vaccination observed between individuals and as a function of physiological stage (Hussen and Schuberth, 2017). By comparing the methylome of purified monocytes, circulating leukocytes and fibroblasts using genome-wide analyses (Jammes et al., 2017), we were able to highlight a specific methylation profile of monocytes affecting genes involved in monocyte function (Figure 5B). To go further, additional analyses need to be performed in order to identify the epigenetic mechanisms that drive the differentiation of monocyte subpopulations.
Figure 5: DNA methylation in immune cells.
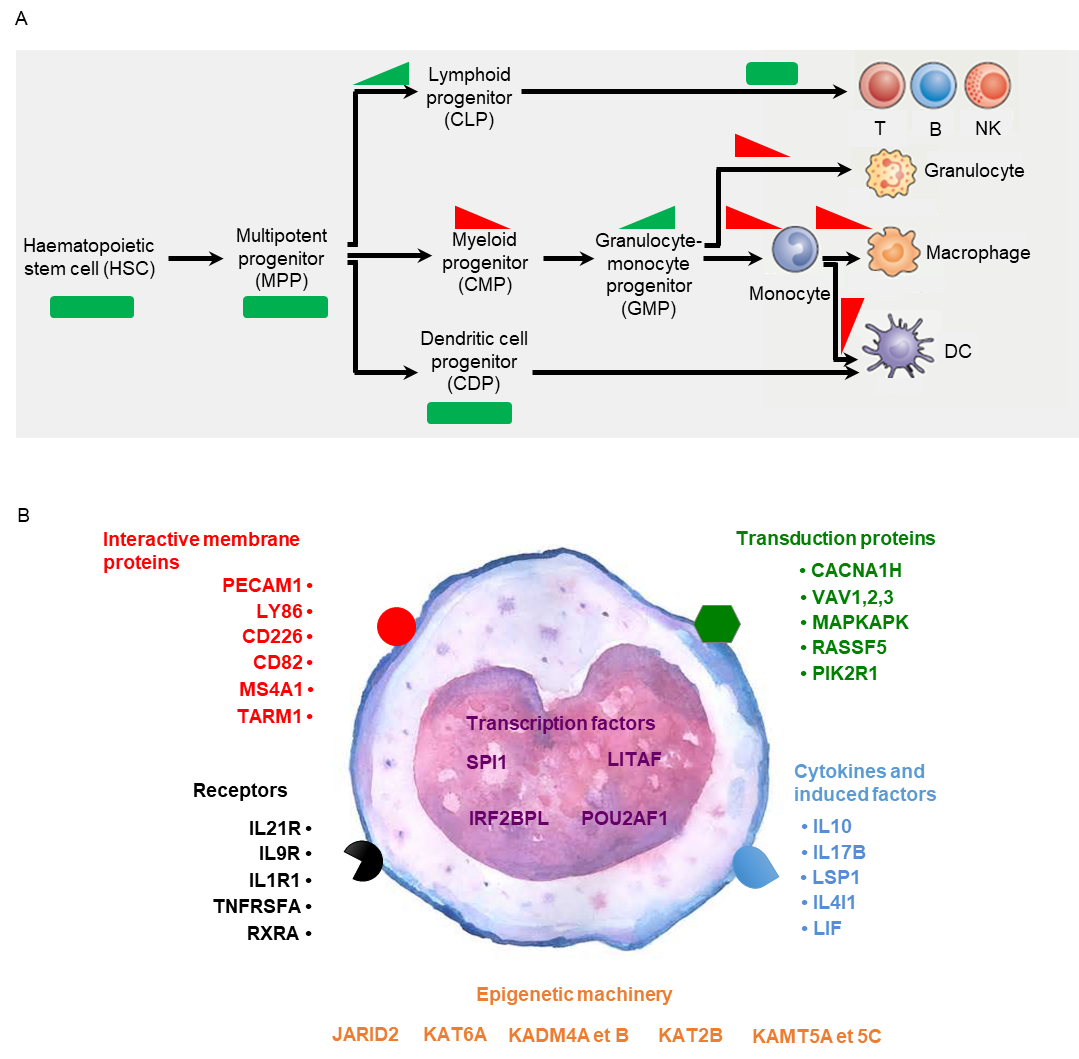
A. Representation of waves of hematopoietic lineage differentiation associated with a gain or loss of 5meC at specific loci (according to Alvarez-Errico et al., 2015). Some stem cells (haematopoietic stem cells, HSC) enter a differentiation phase by generating MPPs (multipotent progenitors), which are divided into 3 subclasses: CLP (common lymphoid progenitors), CMP (common myeloid progenitors) and CDP (common dendritic progenitors) according to the methylation profile acquired under the action of DNMT3A/B for CLP (methylation gain), and TET for CMP (methylation loss). The differences in methylation that characterise subsequent differentiation steps may be significant (monocytes, macrophages and granulocytes) or subtle (B and T lymphocytes, NK cells).
B. Bovine genes displaying a specific methylation profile in monocytes and involved in monocyte differentiation and function (Jammes et al., 2017).
3.2. Epigenetics of the immune response
In dairy cows, mammary infections or mastitis are mainly caused by two pathogens: Escherichia coli and Staphylococcus aureus. The systemic administration of LPS, (liposaccharide), a major component of the E. coli membrane, induces the first stages in the inflammatory reaction involving the mediators and cell types of the innate immune response. In cattle, in vivo exposure to LPS induces the differential expression of genes encoding enzymes of the epigenetic machinery in circulating leukocytes (Doherty et al., 2013). In vitro, the exposure of fibroblasts to LPS causes an increase in the expression of pro-inflammatory cytokine genes. This response is reduced in the presence of an inhibitor of histone deacetyltransferases (Green and Kerr, 2014), which, combined with inhibitors of DNA methylation, also markedly limits inter-individual variability in the response to LPS. These results showed that epigenetic processes are involved in the innate immune response.
Staphylococcus aureus induces subclinical mastitis that is difficult to detect. Transcriptomic variations have been observed in leukocytes from infected dairy cows (61 genes differentially expressed in response to the pathogen) (He et al., 2016). In particular, an increase in the expression of CD4 and IL10 genes involved in the immune response is associated with a decrease in the level of the H3K27me3 repressive mark upstream of the transcription initiation site for these genes.
Resistance to bovine tuberculosis caused by infection with Mycobacterium bovis requires an adaptive immune response mediated by CD4+ lymphocytes. DNA methylation contributes directly to modulating the expression of certain genes mediating the response to infection in CD4+ cells (Doherty et al., 2016), but does not appear to be involved in the response to infection of pulmonary macrophages (O'Doherty et al., 2019). Characterisation of the methylomes of the different subpopulations of immune cells and their variations (inter-individual, following exposure to pathogens, after vaccination) might therefore enable a better estimate of the immune capacity of cattle.
3.3. Epigenetic biomarkers
In humans, numerous studies have demonstrated the possibility of identifying differentially methylated positions in the genome of blood cells in the case of non-immune pathologies (cancers, diabetes, autism) and as a function of lifestyle and/or exposure to toxins, xenobiotics or stress (Dor and Cedar, 2018). Thus, in cattle, the methylome of circulating leukocytes could also be used as a systemic snapshot reflecting the health and life history of individuals. The global circulating leukocyte 5meC levels rise slightly but significantly during the first two months of lactation without any major changes to the proportions of the different immune cell subpopulations (Gasselin et al., 2020). It is therefore necessary to continue a detailed analysis of the genomic distribution of epigenetic modifications in order to identify the molecular processes involved and to identify biomarkers for the physiological status of lactating cows.
Sevane et al. (2019) recently analysed the blood methylome of breeds from the new and old worlds (American breeds and three Iberian breeds), comparing current Iberian bulls with Colombian Creole bulls descending from the founders initially imported from the Iberian Peninsula by Christopher Columbus. The regions differentially methylated in breeds from the new and old worlds are associated with genes involved in the immune response, nervous system functions, energy metabolism, heat resistance, skin and coat colour, suggesting a plasticity of these functions and adaptation to extreme conditions. This pilot study offers a starting point for the identification of epigenetic biomarkers for resilience, which it would be interesting to compare with genetic variations between breeds.
We therefore propose exploring the epigenetic variability of the immune system as a new phenotype for cattle selection, with the aim of obtaining robust and efficient animals whose performance is maintained despite climate changes that might alter the nature and frequency of exposure to pathogens.
4. Milk production
4.1. DNA methylation in the mammary gland
The mammary gland is an organ that undergoes cycles of growth, differentiation and regression during periods of puberty, gestation, lactation and involution after drying out. During lactation, milk synthesis by the mammary epithelial cells is dependent on the expression of numerous genes; environmental factors related to management (milking frequency, diet) or animal health have an impact on this expression and on milk production levels (Dessauge et al., 2011; Boutinaud et al., 2013). Epigenetic regulations may also contribute to regulating milk production. Indeed, studies carried out in rodents or lagomorphs have shown that a combination of epigenetic events (chromatin opening, formation of chromatin loops anchored in the nuclear matrix, histone acetylation, DNA methylation) on a global scale or at the level of certain genes occurs during development of the mammary gland and underpins gene expression (Rijnkels et al., 2010).
In the bovine mammary gland, a distal regulatory region upstream of the casein alpha S1 gene (CSN1S1) is important for its regulation as a function of developmental stage or environmental factors. This region contains binding sites for the STAT-5 transcription factor involved in transduction of the prolactin hormone signal (Vanselow et al., 2006). We showed that this region was less methylated during lactation than during puberty or gestation, and that reducing the milking frequency to one per day induced an increase in methylation associated with a reduction in CSN1S1 expression. Interestingly, an increase in global methylation in half udders producing less milk after one milking per day was also observed (at this stage, breast tissue contains more than 80% mammary epithelial cells; Figure 6) (Nguyen et al., 2014). Even if a trend towards an increase in global methylation was observed, methylation and CSN1S1 expression were not significantly affected by a feed restriction at the onset of lactation (hay and grass silage-based ration compared to one based on dehydrated maize silage and alfalfa supplemented with 30% concentrate; Dessauge et al., 2011). However, the feed restriction during this study was responsible for a 38% fall in milk production and casein K levels (Boutinaud et al., 2016).
Other factors, such as udder health, can modify these epigenetic marks. Escherichia coli infection induces increased CSN1S1 methylation and chromatin condensation associated with decreased expression (Vanselow et al., 2006), while a region in the CXCR1 gene coding for a pro-inflammatory cytokine, and normally hypermethylated in the healthy mammary gland, is demethylated in Staphylococcus aureus-induced mastitis (Mao et al., 2015). Finally, a study was carried out in cloned and contemporary control cows which were reared under the same conditions and produced approximately the same quantity of milk, but with a slightly different composition. The cloned cows appeared to have a particular epigenetic status related to the long-term effects of nuclear reprogramming. Indeed, despite similar 5meC levels in the mammary gland, DNMT1 expression was higher in clones than in control cows (Montazer-Torbati et al., 2016).
Figure 6. Global DNA methylation in the mammary gland and different cell types collected during three studies in dairy cows.
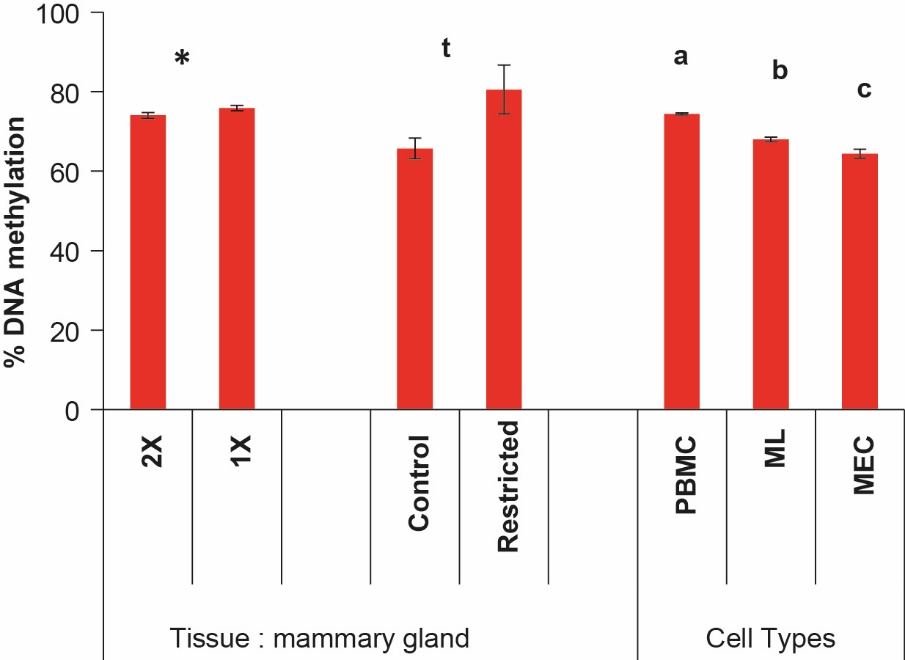
Mammary tissue collected from primiparous cows (n=7) whose udders were milked twice (2X) or once (1X) per day (Nguyen et al., 2014) and from multiparous cows (n=7) fed either a ration meeting 100% of their needs (Control) or a restricted diet (Restricted) during the first 11 weeks of lactation (Boutinaud et al., 2016). Blood (PBMC) and milk (ML) leukocytes and milk mammary epithelial cells (MEC) were collected from primiparous and multiparous dairy cows (n=24); (Gasselin et al., 2020). Histograms represent means per treatment ± SEM. * for P <0.05; t for P = 0.07 and a, b, c for significantly different values, P <0.05. Interestingly, the MEC and mammary gland displayed the same global methylation rate.
4.2 MicroRNAs in the mammary gland and milk
a. miRNA in the mammary gland
Data on the first mammary miRNomes in cattle were published as from 2012 (Li et al., 2012b), and enabled the identification of new miRNAs, some of which are specific to this species (Le Guillou et al., 2014). All the miRNAs described in ruminants are listed in the specific RumimiR database (Bourdon et al., 2019). Studies on the regulation of miRNA expression in dairy cows have highlighted miRNAs specific to lactation stage (Le Guillou et al., 2014), and miRNAs whose expression differs according to breed (Billa et al., 2019) and diet (Mobuchon et al., 2017).
The functions of miRNAs in the bovine mammary gland have so far been little described. Modulations of their expression offer interesting avenues for investigation to understand their role in regulating the expression of genes involved in mammary gland functions. Thus, miR-15a, miR-21-3p, miR-139 and miR-221 regulate the expression of genes involved in the viability or proliferation of mammary epithelial cells; miR-16a, miR-21-3p, miR-27a, miR-34b, miR-130a, miR-181a, miR-224 and miR-454 target genes involved in triglyceride synthesis and milk lipid metabolism, and miR-15a, miR-139 and miR-486 are involved in the synthesis of caseins and other milk constituents. The miRNAs from the mammary gland have also been considered to be of therapeutic interest. A set of miRNAs targeting the gene coding for β-lactoglobulin, one of the major milk allergens, was used to produce a transgenic animal secreting milk that was better tolerated (Jabed et al., 2012).
b. miRNA in milk
Free miRNAs and miRNAs protected in microvesicles or by proteins with which they are associated are present in biological fluids. MicroRNAs are intrinsic and stable components of cow's milk, the majority of which remain after processing procedures such as pasteurisation. MicroRNAs that are present at high levels in total milk (Chen et al., 2010) or in the lipid fraction (Lago Novais et al., 2016) could thus constitute a non-invasive and easily exploitable source of putative biomarkers. A comparison of milk miRNomes from Holstein and Normande cows showed that the proportions of particular miRNAs differed between these two breeds (Le Guillou et al., 2019).
Studies are still necessary to clarify the mechanisms of action and functions related to milk miRNAs. The amount of miRNA absorbed by someone consuming cow's milk indeed appears to be compatible with biological activity (Baier et al., 2014). Moreover, bovine miRNAs may enter peripheral mononuclear cells and affect their gene expression. It has also been shown in vitro that exosomes of bovine milk, carriers of miRNAs, could enter intestinal cells by endocytosis (Wolf et al., 2015). However, the transfer of miRNAs from milk, and their potential impact on the health of the young or general consumers, is currently a controversial subject. For example, our study based on a mouse model producing milk highly enriched in miR-30b did not enable us to demonstrate the transfer of miR-30b to pups receiving this milk (Laubier et al., 2015).
c. Impact of miRNAs on the regulation of epigenetic mechanisms
The overexpression of miR-152 in mammary epithelial cells causes a sharp reduction in the expression of DNMT1 and global levels of 5meC (Wang et al., 2014), while the inhibition of miR-29 expression causes an increase in methylation of the promoters of genes linked to the biosynthesis of milk constituents (Bian et al., 2015). Furthermore, miR-29 and miR-148, which are present in the mammary gland and milk, repress the expression of DNMT3 and DNMT1, respectively, and reduce DNA methylation in humans (Melnik and Schmitz, 2017).
d. Links between polymorphisms and miRNAs involved in milk production traits
Several cases of sequence polymorphisms that can disrupt the miRNA/mRNA interaction and could serve as markers for genomic selection have been described in cattle. For example, a polymorphism in the HMGB1 gene known to play a major role in the innate immune response in the mammary gland alters the miR-223-3p binding site (Li et al., 2012a). Cows with this polymorphism have a stronger expression of HMGB1 and are thought to be less susceptible to mastitis. Genetic variants of miRNA expressed in the mammary gland during lactation and which are located in dairy and mastitis QTLs have been identified (Jiang et al., 2019). The impact of these polymorphisms on the regulatory function of some miRNAs was demonstrated in vitro very recently (Bourdon et al., 2020).
Conclusion
Although epigenetic research started later in cattle than in humans or model species, the number of studies is increasing exponentially as judged by the resulting volume of publications. At INRAE, the production of cloned cattle has provided foundations for pioneering work in epigenetics. Cloning, which induces epigenetic variability with a constant genome, has been and remains a very good model to demonstrate the role of epigenetics in phenotype construction. The examples of experimental work presented in this review, whether based on cloning or not, show that all stages of phenotype construction are based on epigenetic processes, including the development of production traits. The potential uses for epigenetic marks as biomarkers are numerous, particular regarding 5meC or miRNAs in blood, circulating cells, semen and milk.
This demonstration achieved, how can we bridge the distance between the laboratory and the farm? We must now consider epigenetic processes as one of many sources of phenotypic variability in order to generalise their use in livestock management. Indeed, farmed cattle possess variable genotypes and are exposed to less controlled environments than in the experimental farms operated by research institutes such as INRAE. However, genotypes can be determined easily at present: the technology exists and is used routinely for genomic selection. It is difficult to measure the impact of the environment, but epigenetic variations may in fact reflect some of the effects of environmental factors. Estimating and taking account of epigenetic variations could contribute to reducing unexplained phenotypic variance and thus improve the accuracy of estimates for a number of traits of interest.
Artificial intelligence approaches are already being studied to integrate all measurable parameters and thus improve phenotype prediction. The epigenetic marks that are integrated in these models are currently measured using "laboratory" technologies that are too costly to consider their application in the field. Moreover, the complexity and multiplicity of epigenetic marks potentially involved in phenotype control makes it impossible to use them in exhaustive approaches. We anticipate that an important step towards precision farming will be taken when standardised technologies enabling the quantification of variations in a subset of epigenetic marks can be deployed at a large scale, at lower cost and on a routine basis.
Acknowledgements
Most of the work referenced and described in this text was funded by APIS-GENE, ANR, the European Union, Groupe Pilardière, the Animal Physiology and Livestock Systems Division (PHASE) and INRAE. The authors would like to thank all members of their respective teams, their collaborators as well as the breeding companies Évolution, Umotest, Evajura, AWE and XR Repro. The authors also give special thanks to Jean-Paul Renard who was a pioneer in research on epigenetics in cattle. The data resulting from work carried out in INRAE laboratories could not have been produced without the involvement of ALLICE staff, platforms and experimental units and facilities. We would like to thank them here.
This article was first translated with www.DeepL.com/Translator and the authors would like to thank Victoria Hawken for English language editing.
References
- Allfrey V.G., Mirsky A.E., 1964. Structural Modifications of Histones and their Possible Role in the Regulation of RNA Synthesis. Science, 144, 559.
- Alvarez-Errico D., Vento-Tormo R., Sieweke M., Ballestar E., 2015. Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol., 15, 7-17.
- Ao X., Sa R., Wang J., Dao R., Wang H., Yu H., 2016. Activation-induced cytidine deaminase selectively catalyzed active DNA demethylation in pluripotency gene and improved cell reprogramming in bovine SCNT embryo. Cytotechnology, 68, 2637-2648.
- Baier S.R., Nguyen C., Xie F., Wood J.R., Zempleni J., 2014. MicroRNAs are absorbed in biologically meaningful amounts from nutritionally relevant doses of cow milk and affect gene expression in peripheral blood mononuclear cells, HEK-293 kidney cell cultures, and mouse livers. J. Nutr., 144, 1495-1500.
- Bayoumi A.S., Sayed A., Broskova Z., Teoh J.P., Wilson J., Su H., Tang Y.L., Kim I.M., 2016. Crosstalk between Long Noncoding RNAs and MicroRNAs in Health and Disease. Int. J. Mol. Sci., 17, 356.
- Beaujean N., 2018. Epigenetic features of animal biotechnologies. In: Niemann H., Wrenzycki C. (Eds). Anim. Biotechnol., 2 Springer, Cham. doi:10.1007/978-3-319-92348-2_3
- Berger S.L., Kouzarides T., Shiekhattar R., Shilatifard A., 2009. An operational definition of epigenetics. Genes Dev., 23, 781-783.
- Bian Y., Lei Y., Wang C., Wang J., Wang L., Liu L., Liu L., Gao X., Li Q., 2015. Epigenetic Regulation of miR-29s Affects the Lactation Activity of Dairy Cow Mammary Epithelial Cells. J. Cell Physiol., 230, 2152-2163.
- Billa P.A., Faulconnier Y., Ye T., Chervet M., Le Provost F., Pires J.A.A., Leroux C., 2019. Deep RNA-Seq reveals miRNome differences in mammary tissue of lactating Holstein and Montbeliarde cows. BMC Genomics, 20, 621.
- Bissonnette N., Levesque-Sergerie J.P., Thibault C., Boissonneault G., 2009. Spermatozoal transcriptome profiling for bull sperm motility: a potential tool to evaluate semen quality. Reproduction, 138, 65-80.
- Boichard D., Ducrocq V., Croiseau P., Fritz S., 2016. Genomic selection in domestic animals: Principles, applications and perspectives. Comptes Rendus Biol., 339, 274-277.
- Boskovic A., Bender A., Gall L., Ziegler-Birling C., Beaujean N., Torres-Padilla M.E., 2012. Analysis of active chromatin modifications in early mammalian embryos reveals uncoupling of H2A.Z acetylation and H3K36 trimethylation from embryonic genome activation. Epigenetics, 7, 747-757.
- Bouquet A., Juga J., 2013. Integrating genomic selection into dairy cattle breeding programmes: a review. Animal, 7, 705-713.
- Bourdon C., Bardou P., Aujean E., Le Guillou S., Tosser-Klopp G., Le Provost F., 2019. RumimiR: a detailed microRNA database focused on ruminant species. Database (Oxford), 2019.
- Bourdon C., Boussaha M., Sanchez M.P., Tribout T., Lefebvre R., Le Guillou S., Tosser-Klopp G., Le Provost F., 2020. Identification and characterization of microRNA genetic variants in dairy cattle, from their detection to the analysis of their biological impacts. In: PAG XXVIII - Plant and Animal Genome Conference. San Diego, USA.
- Boutinaud M., Galio L., Lollivier V., Finot L., Wiart S., Esquerre D., Devinoy E., 2013. Unilateral once daily milking locally induces differential gene expression in both mammary tissue and milk epithelial cells revealing mammary remodeling. Physiol Genomics, 45, 973-985.
- Boutinaud M., Lollivier V., Dessauge F., Devinoy E., Singh K., 2016. Régulations épigénétiques et activation de la transcription lors d’une restriction alimentaire chez la vache laitière. In: Journées d’Animation des Crédits Incitatifs du Département de Physiologie Animale et Systèmes d’Elevage (JACI Phase 2016). Tours, France.
- Bushati N., Cohen S.M., 2007. microRNA functions. Annu. Rev. Cell Dev. Biol., 23, 175-205.
- Byrne C.J., Fair S., English A.M., Cirot M., Staub C., Lonergan P., Kenny D.A., 2018. Plane of nutrition before and after 6 months of age in Holstein-Friesian bulls: I. Effects on performance, body composition, age at puberty, and postpubertal semen production. J. Dairy Sci., 101, 3447-3459.
- Camargo L.S.A., Aguirre-Lavin T., Adenot P., Araujo T.D., Mendes V.R.A., Louro I.D., Beaujean N., Souza E.D., 2019. Heat shock during in vitro maturation induces chromatin modifications in the bovine embryo. Reproduction.
- Carrell D.T., 2012. Epigenetics of the male gamete. Fertil. Steril., 97, 267-274.
- Chen X., Gao C., Li H., Huang L., Sun Q., Dong Y., Tian C., Gao S., Dong H., Guan D., Hu X., Zhao S., Li L., Zhu L., Yan Q., Zhang J., Zen K., Zhang C.Y., 2010. Identification and characterization of microRNAs in raw milk during different periods of lactation, commercial fluid, and powdered milk products. Cell Res., 20, 1128-1137.
- Choudhry H., Catto J.W., 2011. Epigenetic regulation of microRNA expression in cancer. Methods Mol. Biol., 676, 165-184.
- Chung N., Bogliotti Y.S., Ding W., Vilarino M., Takahashi K., Chitwood J.L., Schultz R.M., Ross P.J., 2017. Active H3K27me3 demethylation by KDM6B is required for normal development of bovine preimplantation embryos. Epigenetics, 12, 1048-1056.
- Dessauge F., Lollivier V., Ponchon B., Bruckmaier R., Finot L., Wiart S., Cutullic E., Disenhaus C., Barbey S., Boutinaud M., 2011. Effects of nutrient restriction on mammary cell turnover and mammary gland remodeling in lactating dairy cows. J. Dairy Sci., 94, 4623-4635.
- Doherty R., O'Farrelly C., Meade K.G., 2013. Epigenetic regulation of the innate immune response to LPS in bovine peripheral blood mononuclear cells (PBMC). Vet. Immunol. Immunopathol., 154, 102-110.
- Doherty R., Whiston R., Cormican P., Finlay E.K., Couldrey C., Brady C., O'Farrelly C., Meade K.G., 2016. The CD4(+) T cell methylome contributes to a distinct CD4(+) T cell transcriptional signature in Mycobacterium bovis-infected cattle. Sci. Rep., 6, 31014.
- Dor Y., Cedar H., 2018. Principles of DNA methylation and their implications for biology and medicine. Lancet, 392, 777-786.
- Fagerlind M., Stalhammar H., Olsson B., Klinga-Levan K., 2015. Expression of miRNAs in Bull Spermatozoa Correlates with Fertility Rates. Reprod. Domest. Anim., 50, 587-594.
- Gasselin M., Boutinaud M., Prezelin A., Debournoux P., Fargetton M., Mariani E., Zawadzki J., Kiefer H., Jammes H., 2020. Effects of micronutrient supplementation on performance and epigenetic status in dairy cows. Animal, 1-10.
- Gkountela S., Zhang K.X., Shafiq T.A., Liao W.W., Hargan-Calvopina J., Chen P.Y., Clark A.T., 2015. DNA Demethylation Dynamics in the Human Prenatal Germline. Cell, 161, 1425-1436.
- Govindaraju A., Uzun A., Robertson L., Atli M.O., Kaya A., Topper E., Crate E.A., Padbury J., Perkins A., Memili E., 2012. Dynamics of microRNAs in bull spermatozoa. Reprod. Biol. Endocrinol., 10, 82.
- Green B.B., Kerr D.E., 2014. Epigenetic contribution to individual variation in response to lipopolysaccharide in bovine dermal fibroblasts. Vet. Immunol. Immunopathol., 157, 49-58.
- Griffiths-Jones S., Saini H.K., van Dongen S., Enright A.J., 2008. miRBase: tools for microRNA genomics. Nucleic Acids Res., 36, D154-158.
- Guillomot M., Prezelin A., Campion E., Kiefer H., Beaujean N., Aguirre-Lavin T., Jammes H., 2014. Analysis of 5-methylcytosine and 5-hydromethylcytosine content and distribution in bovine placental and fetal tissues after somatic nuclear reprogramming. In: Int.Fed. Placenta Assoc. Meet. Placenta (Ed). Elsevier, Paris, France, A36-A37.
- Guo F., Yan L., Guo H., Li L., Hu B., Zhao Y., Yong J., Hu Y., Wang X., Wei Y., Wang W., Li R., Yan J., Zhi X., Zhang Y., Jin H., Zhang W., Hou Y., Zhu P., Li J., Zhang L., Liu S., Ren Y., Zhu X., Wen L., Gao Y.Q., Tang F., Qiao J., 2015. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells. Cell, 161, 1437-1452.
- He Y., Song M., Zhang Y., Li X., Song J., Zhang Y., Yu Y., 2016. Whole-genome regulation analysis of histone H3 lysin 27 trimethylation in subclinical mastitis cows infected by Staphylococcus aureus. BMC Genomics, 17, 565.
- Heyman Y., 2005. Nuclear transfer: a new tool for reproductive biotechnology in cattle. Reprod. Nutr. Dev., 45, 353-361.
- Hill P.W.S., Leitch H.G., Requena C.E., Sun Z., Amouroux R., Roman-Trufero M., Borkowska M., Terragni J., Vaisvila R., Linnett S., Bagci H., Dharmalingham G., Haberle V., Lenhard B., Zheng Y., Pradhan S., Hajkova P., 2018. Epigenetic reprogramming enables the transition from primordial germ cell to gonocyte. Nature, 555, 392-396.
- Hussen J., Schuberth H.J., 2017. Heterogeneity of Bovine Peripheral Blood Monocytes. Front. Immunol., 8, 1875.
- Jabed A., Wagner S., McCracken J., Wells D.N., Laible G., 2012. Targeted microRNA expression in dairy cattle directs production of beta-lactoglobulin-free, high-casein milk. Proc. Natl. Acad. Sci., U S A, 109, 16811-16816.
- Jammes H., Gasselin M., Perrier J.P., Piumi F., Jouneau L., Al Adhami H., Prezelin A., Boutinaud M., Leroux C., Pires J., Weber M., Pount B., Jzawadski J., Kiefer H., 2017. Methylation analysis in monocytes at postpartum period in dairy cattle. In: PAG XXV - Plant and Animal Genome Conference. San Diego, USA.
- Jenuwein T., 2006. The epigenetic magic of histone lysine methylation. FEBS J., 273, 3121-3135.
- Jiang Q., Zhao H., Li R., Zhang Y., Liu Y., Wang J., Wang X., Ju Z., Liu W., Hou M., Huang J., 2019. In silico genome-wide miRNA-QTL-SNPs analyses identify a functional SNP associated with mastitis in Holsteins. BMC Genet., 20, 46.
- Jiang Z., Lin J., Dong H., Zheng X., Marjani S.L., Duan J., Ouyang Z., Chen J., Tian X.C., 2018. DNA methylomes of bovine gametes and in vivo produced preimplantation embryos. Biol. Reprod., 99, 949-959.
- Junien C., Panchenko P., Fneich S., Pirola L., Chriett S., Amarger V., Kaeffer B., Parnet P., Torrisani J., Bolanos Jimenez F., Jammes H., Gabory A., 2016. Epigenetics in transgenerational responses to environmental impacts: from facts and gaps. Med. Sci., 32, 35-44.
- Kiefer H., Jouneau L., Campion E., Rousseau-Ralliard D., Larcher T., Martin-Magniette M.L., Balzergue S., Ledevin M., Prezelin A., Chavatte-Palmer P., Heyman Y., Richard C., Le Bourhis D., Renard J.P., Jammes H., 2016. Altered DNA methylation associated with an abnormal liver phenotype in a cattle model with a high incidence of perinatal pathologies. Sci. Rep., 6, 38869.
- Kiefer H., Perrier J.P., 2019. DNA methylation in bull spermatozoa: evolutionary impacts, interindividual variability, and contribution to the embryo. In: Sci. C.J.o.A. (Ed), CSAS Symp. 2018 American Soc. Anim. Sci. (ASAS) and Canadian Soc. Anim. Sci. (CSAS) Annual Meeting and Trade Show. NRC Research Press, Vancouver, Canada.
- Kouzarides T., 2007. Chromatin modifications and their function. Cell, 128, 693-705.
- Lago Novais D., Pawlowski K., Pires J., Mobuchon L., Bes S., Leroux C., 2016. Milk fat globules as a source of mammary microRNA. In: 2016 JAM, Joint Ann. Meet., "Animals and Science: Big Solutions for Grand Challenges". Science J.o.A. (Ed). ASAS – Ame. Soc. Anim. Sci., Salt Lake City, USA, 401.
- Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T., 2002. Identification of tissue-specific microRNAs from mouse. Curr. Biol., 12, 735-739.
- Lambert S., Blondin P., Vigneault C., Labrecque R., Dufort I., Sirard M.A., 2018. Spermatozoa DNA methylation patterns differ due to peripubertal age in bulls. Theriogenology, 106, 21-29.
- Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A.O., Landthaler M., Lin C., Socci N.D., Hermida L., Fulci V., Chiaretti S., Foa R., Schliwka J., Fuchs U., Novosel A., Muller R.U., Schermer B., Bissels U., Inman J., Phan Q., Chien M., Weir D.B., Choksi R., De Vita G., Frezzetti D., Trompeter H.I., Hornung V., Teng G., Hartmann G., Palkovits M., Di Lauro R., Wernet P., Macino G., Rogler C.E., Nagle J.W., Ju J., Papavasiliou F.N., Benzing T., Lichter P., Tam W., Brownstein M.J., Bosio A., Borkhardt A., Russo J.J., Sander C., Zavolan M., Tuschl T., 2007. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell, 129, 1401-1414.
- Laubier J., Castille J., Le Guillou S., Le Provost F., 2015. No effect of an elevated miR-30b level in mouse milk on its level in pup tissues. RNA Biol., 12, 26-29.
- Le Guillou S., Marthey S., Laloe D., Laubier J., Mobuchon L., Leroux C., Le Provost F., 2014. Characterisation and comparison of lactating mouse and bovine mammary gland miRNomes. PLoS One, 9, e91938.
- Le Guillou S., Leduc A., Laubier J., Barbey S., Rossignol M.N., Lefebvre R., Marthey S., Laloe D., Le Provost F., 2019. Characterization of Holstein and Normande whole milk miRNomes highlights breed specificities. Sci. Rep., 9, 20345.
- Li L., Huang J., Zhang X., Ju Z., Qi C., Zhang Y., Li Q., Wang C., Miao W., Zhong J., Hou M., Hang S., 2012a. One SNP in the 3'-UTR of HMGB1 gene affects the binding of target bta-miR-223 and is involved in mastitis in dairy cattle. Immunogenetics, 64, 817-824.
- Li Z., Liu H., Jin X., Lo L., Liu J., 2012b. Expression profiles of microRNAs from lactating and non-lactating bovine mammary glands and identification of miRNA related to lactation. BMC Genomics, 13, 731.
- Ly L., Chan D., Trasler J.M., 2015. Developmental windows of susceptibility for epigenetic inheritance through the male germline. Semin. Cell Dev. Biol., 43, 96-105.
- Lyko F., 2018. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet., 19, 81-92.
- Mandon-Pépin B., Allais-Bonnet A., André M., Calvel P., Sellem E., Le Danvic C., Prézelin A., Jammes H., Pailhoux E., Kiefer H., Pannetier M., 2017. Epigenetic reprogramming in the male germ cells of ruminants. In: France Japan Epigenetics Workshop. Paris, France.
- Mao Y.J., Zhu X.R., Li R., Chen D., Xin S.Y., Zhu Y.H., Liao X.X., Wang X.L., Zhang H.M., Yang Z.P., Yang L.G., 2015. Methylation analysis of CXCR1 in mammary gland tissue of cows with mastitis induced by Staphylococcus aureus. Genet. Mol. Res., 14, 12606-12615.
- Melnik B.C., Schmitz G., 2017. MicroRNAs: Milk's epigenetic regulators. Best Pract Res Clin Endocrinol. Metab., 31, 427-442.
- Mobuchon L., Le Guillou S., Marthey S., Laubier J., Laloe D., Bes S., Le Provost F., Leroux C., 2017. Sunflower oil supplementation affects the expression of miR-20a-5p and miR-142-5p in the lactating bovine mammary gland. PLoS One, 12, e0185511.
- Montazer-Torbati F., Boutinaud M., Brun N., Richard C., Neveu A., Jaffrezic F., Laloe D., LeBourhis D., Nguyen M., Chadi S., Jammes H., Renard J.P., Chat S., Boukadiri A., Devinoy E., 2016. Differences during the first lactation between cows cloned by somatic cell nuclear transfer and noncloned cows. J. Dairy Sci., 99, 4778-4794.
- Monteys A.M., Spengler R.M., Wan J., Tecedor L., Lennox K.A., Xing Y., Davidson B.L., 2010. Structure and activity of putative intronic miRNA promoters. RNA, 16, 495-505.
- Mujtaba S., Zeng L., Zhou M.M., 2007. Structure and acetyl-lysine recognition of the bromodomain. Oncogene, 26, 5521-5527.
- Nguyen M., Boutinaud M., Petridou B., Gabory A., Pannetier M., Chat S., Bouet S., Jouneau L., Jaffrezic F., Laloe D., Klopp C., Brun N., Kress C., Jammes H., Charlier M., Devinoy E., 2014. DNA methylation and transcription in a distal region upstream from the bovine AlphaS1 casein gene after once or twice daily milking. PLoS One, 9, e111556.
- Nishibuchi G., Dejardin J., 2017. The molecular basis of the organization of repetitive DNA-containing constitutive heterochromatin in mammals. Chromosome Res., 25, 77-87.
- O'Doherty A.M., O'Shea L.C., Fair T., 2012. Bovine DNA methylation imprints are established in an oocyte size-specific manner, which are coordinated with the expression of the DNMT3 family proteins. Biol. Reprod., 86, 67.
- O'Doherty A.M., Rue-Albrecht K.C., Magee D.A., Ahting S., Irwin R.E., Hall T.J., Browne J.A., Nalpas N.C., Walsh C.P., Gordon S.V., Wojewodzic M.W., MacHugh D.E., 2019. The bovine alveolar macrophage DNA methylome is resilient to infection with Mycobacterium bovis. Sci. Rep., 9, 1510.
- Perrier J.P., Kenny D.A., Chaulot-Talmon A., Byrne C.J., Sellem E., Jouneau L., Aubert-Frambourg A., Schibler L., Jammes H., Lonergan P., Fair S. and Kiefer H., 2020. Accelerating onset of puberty through modification of early life nutrition induces modest but persistent changes in bull sperm DNA methylation profiles post-puberty. Front. Genet., 11, 945.
- Perrier J.P., Sellem E., Jouneau L., Boussaha M., Hozé C., Fritz S., Aubert-Frambourg A., Le Danvic C., Boichard D., Schibler L., Jammes H., Kiefer H., 2019. Genetic and non-genetic factors determine DNA methylation patterns in bull sperm. In: Annual meeting of the European Association for Animal Production (EAAP). (EAAP) E.F.o.A.S. (Ed), Ghent, Belgium, 48.
- Perrier J.P., Sellem E., Prezelin A., Gasselin M., Jouneau L., Piumi F., Al Adhami H., Weber M., Fritz S., Boichard D., Le Danvic C., Schibler L., Jammes H., Kiefer H., 2018. A multi-scale analysis of bull sperm methylome revealed both species peculiarities and conserved tissue-specific features. BMC Genomics, 19, 404.
- Pichugin A., Le Bourhis D., Adenot P., Lehmann G., Audouard C., Renard J.P., Vignon X., Beaujean N., 2010. Dynamics of constitutive heterochromatin: two contrasted kinetics of genome restructuring in early cloned bovine embryos. Reproduction, 139, 129-137.
- Radford E.J., Ito M., Shi H., Corish J.A., Yamazawa K., Isganaitis E., Seisenberger S., Hore T.A., Reik W., Erkek S., Peters A., Patti M.E., Ferguson-Smith A.C., 2014. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science, 345, 1255903.
- Rijnkels M., Kabotyanski E., Montazer-Torbati M.B., Hue Beauvais C., Vassetzky Y., Rosen J.M., Devinoy E., 2010. The epigenetic landscape of mammary gland development and functional differentiation. J. Mammary Gland Biol. Neoplasia, 15, 85-100.
- Ross P.J., Sampaio R.V., 2018. Epigenetic remodeling in preimplantation embryos: cows are not big mice. In: Reproduction A. (Ed), 32nd Ann. Meet. Brazilian Embryo Technol. Soc. (SBTE), Florianopólis, Brazil, 204-2014.
- Schubeler D., 2015. Function and information content of DNA methylation. Nature, 517, 321-326.
- Seisenberger S., Andrews S., Krueger F., Arand J., Walter J., Santos F., Popp C., Thienpont B., Dean W., Reik W., 2012. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell., 48, 849-862.
- Sellem E., Marthey S., Rau A., Jouneau L., Bonnet A., Perrier J.P., Fritz S., Le Danvic C., Boussaha M., Kiefer H., Jammes H., Schibler L., 2020. A comprehensive overview of bull sperm-borne small non-coding RNAs and their diversity across breeds. Epigenetics Chromatin, 13, 19.
- Sevane N., Martinez R., Bruford M.W., 2019. Genome-wide differential DNA methylation in tropically adapted Creole cattle and their Iberian ancestors. Anim. Genet., 50, 15-26.
- Stinshoff H., Wilkening S., Hanstedt A., Bruning K., Wrenzycki C., 2011. Cryopreservation affects the quality of in vitro produced bovine embryos at the molecular level. Theriogenology, 76, 1433-1441.
- Takeda K., Kobayashi E., Nishino K., Imai A., Adachi H., Hoshino Y., Iwao K., Akagi S., Kaneda M., Watanabe S., 2019. Age-related changes in DNA methylation levels at CpG sites in bull spermatozoa and in vitro fertilization-derived blastocyst-stage embryos revealed by combined bisulfite restriction analysis. J. Reprod. Dev., 65, 305-312.
- Vanselow J., Yang W., Herrmann J., Zerbe H., Schuberth H.J., Petzl W., Tomek W., Seyfert H.M., 2006. DNA-remethylation around a STAT5-binding enhancer in the alphaS1-casein promoter is associated with abrupt shutdown of alphaS1-casein synthesis during acute mastitis. J. Mol. Endocrinol., 37, 463-477.
- Wang J., Bian Y., Wang Z., Li D., Wang C., Li Q., Gao X., 2014. MicroRNA-152 regulates DNA methyltransferase 1 and is involved in the development and lactation of mammary glands in dairy cows. PLoS One, 9, e101358.
- Weber J.A., Baxter D.H., Zhang S., Huang D.Y., Huang K.H., Lee M.J., Galas D.J., Wang K., 2010. The microRNA spectrum in 12 body fluids. Clin. Chem., 56, 1733-1741.
- Wolf T., Baier S.R., Zempleni J., 2015. The Intestinal Transport of Bovine Milk Exosomes Is Mediated by Endocytosis in Human Colon Carcinoma Caco-2 Cells and Rat Small Intestinal IEC-6 Cells. J. Nutr., 145, 2201-2206.
- Wu X., Zhang Y., 2017. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet., 18, 517-534.
Abstract
In cattle breeding and farming, epigenetic mechanisms are regarded as a potential source of phenotypic variance unaccounted by genetic selection and could be used to better control the environmental impact on the genetic potential of animals. In this review, we briefly describe epigenetic processes, and address, from research studies mainly conducted at INRAE, the major role of epigenetic factors in the elaboration of phenotype in cattle. Focus is given to environmental factors shaping the epigenome, leading to variable effects on fertility, development, health and dairy production. The potential for using epigenetic markers in precision farming and as a complementary approach to genomic selection is also discussed.
Attachments
No supporting information for this articleArticle statistics
 Views: 17265
Views: 17265
Downloads
 XML: 141
XML: 141
 PDF: 911
PDF: 911